Department of Cell Biology and Physiology, and Center for the Investigation of Membrane Excitability Diseases, Washington University School of Medicine, St. Louis, Missouri, United States of America.
PLoS One. 2013 May 7;8(5):e63758. doi: 10.1371/journal.pone.0063758. Print 2013.
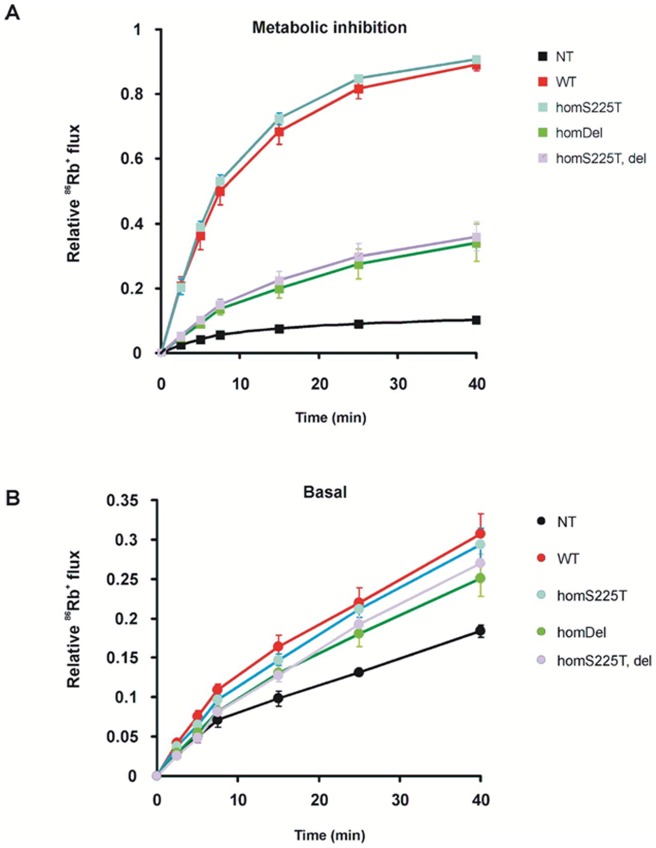
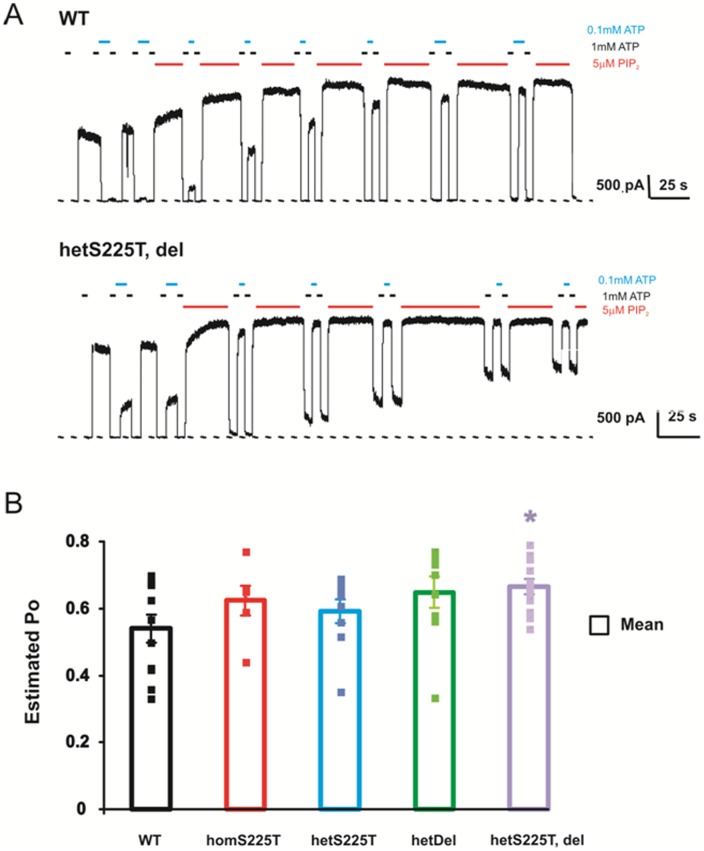
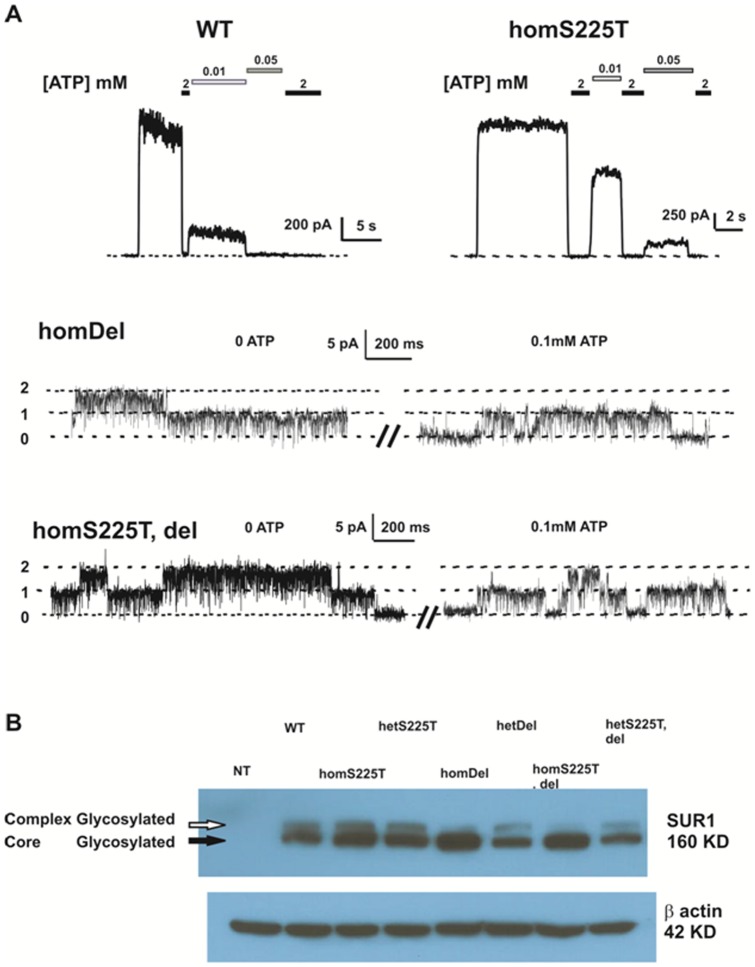
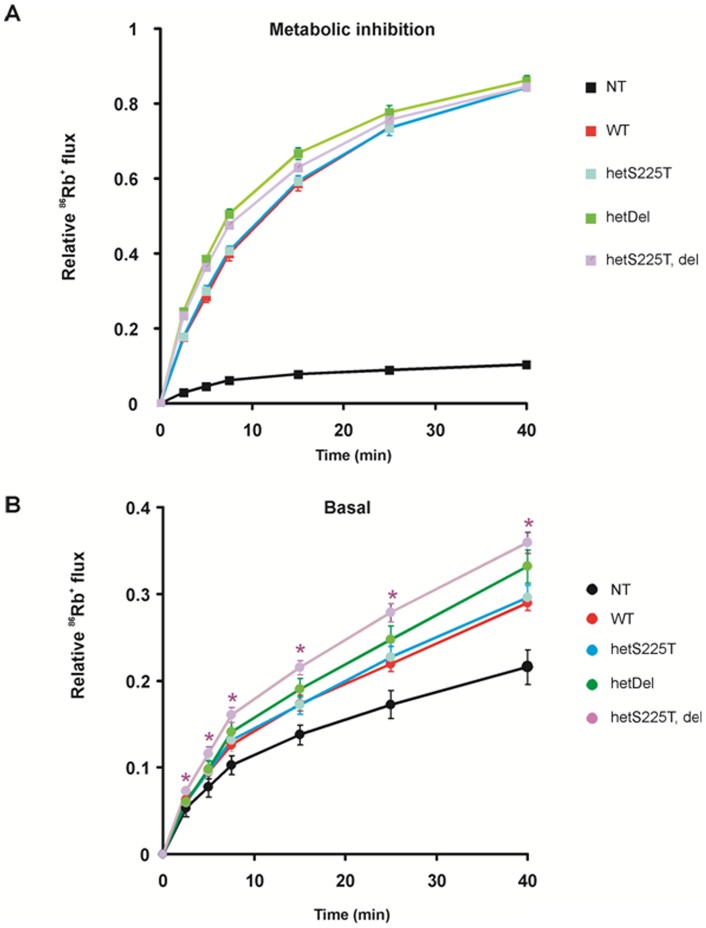
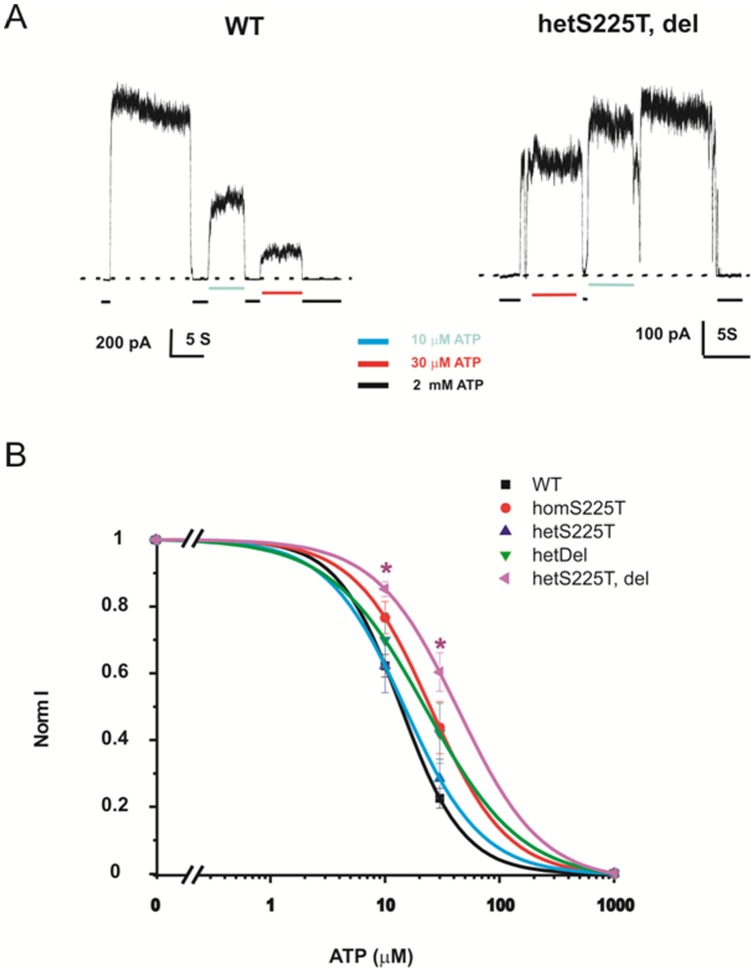
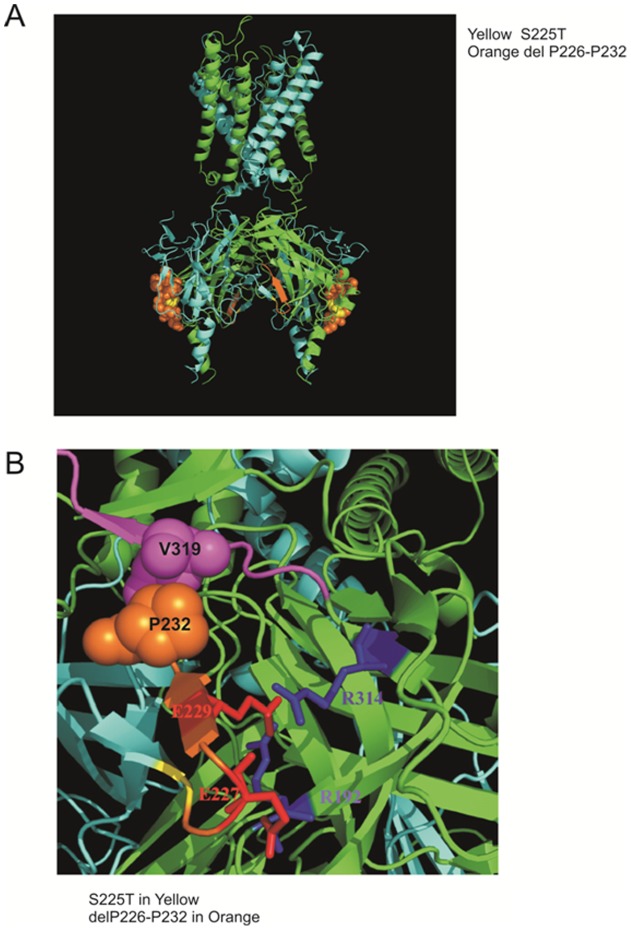
ATP-sensitive potassium (K(ATP)) channels are widely distributed in various tissues and cell types where they couple cell metabolism to cell excitability. Gain of channel function (GOF) mutations in the genes encoding Kir6.2 (KCNJ11) or the associated regulatory ssulfonylurea receptor 1 subunit (ABCC8), cause developmental delay, epilepsy and neonatal diabetes (DEND) due to suppressed cell excitability in pancreatic β-cells and neurons. The objective of this study was to determine the molecular basis of infancy-onset diabetes and a mild form of intermediate DEND, resulting from a novel KCNJ11 in frame mutation plus deletion. The naturally occurring Kir6.2 mutation plus deletion (Ser225Thr, Pro226_Pro232del) as well as the isolated S225T mutation or isolated del226-232 deletion were coexpressed with SUR1 in COS cells in homozygous or heterozygous states. The protein expression and gating effects of the resulting channels were assessed biochemically and electrophysiologically. For both the deletion and point mutations, simulated heterozygous expression resulted in overall increased conductance in intact cells in basal conditions and rightward shifted ATP dose-response curves in excised patches, due to increased intrinsic open probability. Interestingly, homomeric channels for the combined deletion/mutation, or for the deletion alone, showed dramatically reduced channel expression at the cell membrane, which would underlie a reduced function in vivo. These results demonstrate that both the mis-sense mutation and the deleted region in the Kir6.2 subunit are important for control of the intrinsic channel gating and suggest that the clinical presentation could be affected by the competition between loss-of-function by reduced trafficking and enhanced channel gating.
三磷酸腺苷敏感性钾 (K(ATP)) 通道广泛分布于各种组织和细胞类型中,它们将细胞代谢与细胞兴奋耦联。编码 Kir6.2 (KCNJ11) 的基因或相关的磺酰脲受体 1 亚基 (ABCC8) 的通道功能获得性 (GOF) 突变导致发育迟缓、癫痫和新生儿糖尿病 (DEND),原因是胰腺 β 细胞和神经元的细胞兴奋受到抑制。本研究的目的是确定婴儿期发病的糖尿病和轻度中间 DEND 的分子基础,这是由于新型 KCNJ11 框移突变加缺失引起的。天然存在的 Kir6.2 突变加缺失 (Ser225Thr,Pro226_Pro232del) 以及分离的 S225T 突变或分离的 del226-232 缺失与 SUR1 在 COS 细胞中以纯合或杂合状态共同表达。通过生化和电生理方法评估所得通道的蛋白表达和门控效应。对于缺失和点突变,模拟杂合表达导致在基础条件下完整细胞的整体电导增加,并且在分离的斑块中 ATP 剂量反应曲线右移,这是由于内在开放概率增加所致。有趣的是,对于组合缺失/突变或单独缺失的同型通道,在细胞膜上显示出明显减少的通道表达,这将是体内功能降低的基础。这些结果表明,Kir6.2 亚基中的错义突变和缺失区域对于控制内在通道门控都很重要,并表明临床表型可能受到减少转运和增强通道门控之间竞争的影响。