Department of Biology, Lund University, Ecology Building, Lund, SE 22362, Sweden.
BMC Genomics. 2013 May 14;14:330. doi: 10.1186/1471-2164-14-330.
Animal migration requires adaptations in morphological, physiological and behavioural traits. Several of these traits have been shown to possess a strong heritable component in birds, but little is known about their genetic architecture. Here we used 454 sequencing of brain-derived transcriptomes from two differentially migrating subspecies of the willow warbler Phylloscopus trochilus to detect genes potentially underlying traits associated with migration.
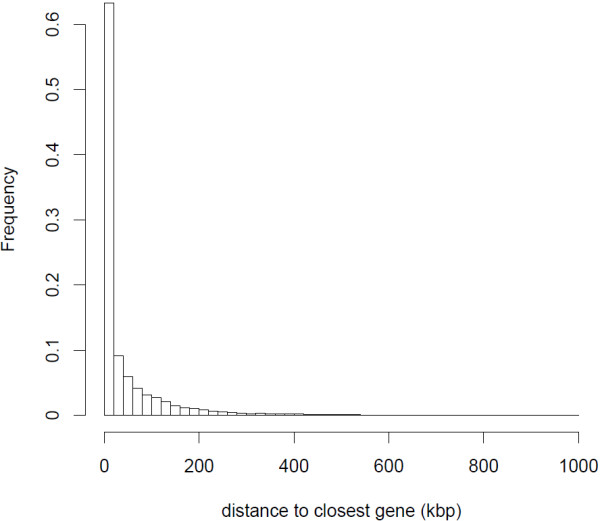
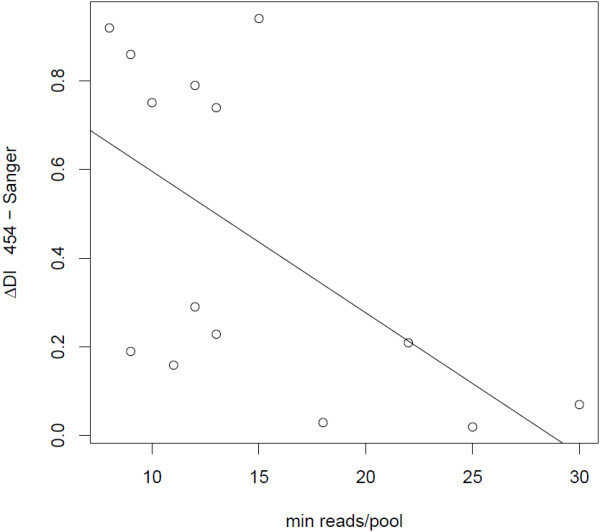
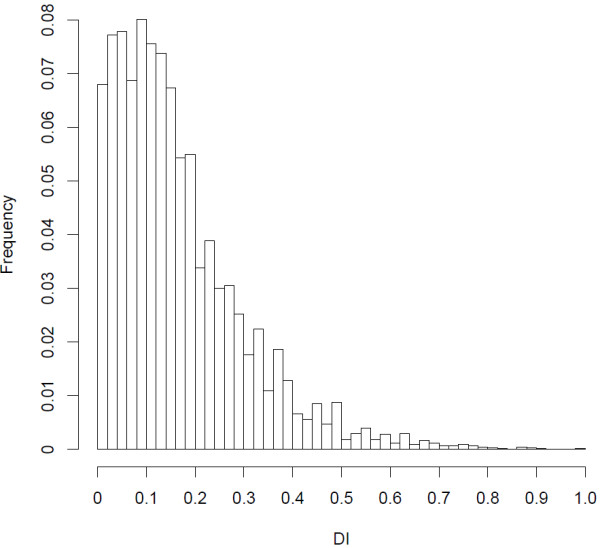
The transcriptome sequencing resulted in 1.8 million reads following filtering steps. Most of the reads (84%) were successfully mapped to the genome of the zebra finch Taeniopygia gutatta. The mapped reads were situated within at least 12,101 predicted zebra finch genes, with the greatest sequencing depth in exons. Reads that were mapped to intergenic regions were generally located close to predicted genes and possibly located in uncharacterized untranslated regions (UTRs). Out of 85,000 single nucleotide polymorphisms (SNPs) with a minimum sequencing depth of eight reads from each of two subspecies-specific pools, only 55 showed high differentiation, confirming previous studies showing that most of the genetic variation is shared between the subspecies. Validation of a subset of the most highly differentiated SNPs using Sanger sequencing demonstrated that several of them also were differentiated between an independent set of individuals of each subspecies. These SNPs were clustered in two chromosome regions that are likely to be influenced by divergent selection between the subspecies and that could potentially be associated with adaptations to their different migratory strategies.
Our study represents the first large-scale sequencing analysis aiming at detecting genes underlying migratory phenotypes in birds and provides new candidates for genes potentially involved in migration.
动物迁徙需要在形态、生理和行为特征上做出适应。这些特征中的许多都在鸟类中表现出很强的遗传成分,但它们的遗传结构知之甚少。在这里,我们使用两种不同迁徙习性的柳莺 Phylloscopus trochilus 的大脑衍生转录组的 454 测序,来检测可能与迁徙相关的特征的潜在基因。
经过过滤步骤,转录组测序得到了 180 万个reads。大多数reads(84%)成功映射到斑胸草雀 Taeniopygia gutatta 的基因组。映射的reads位于至少 12101 个预测的斑胸草雀基因内,外显子中的测序深度最大。映射到基因间区的reads通常靠近预测基因,可能位于未表征的非翻译区(UTR)中。在两个亚种特异性池的每个池至少有 8 个reads 的 85000 个单核苷酸多态性(SNP)中,只有 55 个表现出高度分化,这证实了之前的研究表明,亚种之间共享大多数遗传变异。使用 Sanger 测序对一组高度分化的 SNP 进行验证,表明其中一些 SNP 在每个亚种的一组独立个体之间也存在分化。这些 SNP 聚集在两个染色体区域,这些区域可能受到亚种之间分歧选择的影响,并且可能与它们不同的迁徙策略的适应有关。
我们的研究代表了首次大规模测序分析,旨在检测鸟类迁徙表型的潜在基因,并为可能参与迁徙的基因提供了新的候选基因。