Romay Maria C, Millard Mark J, Glaubitz Jeffrey C, Peiffer Jason A, Swarts Kelly L, Casstevens Terry M, Elshire Robert J, Acharya Charlotte B, Mitchell Sharon E, Flint-Garcia Sherry A, McMullen Michael D, Holland James B, Buckler Edward S, Gardner Candice A
Genome Biol. 2013 Jun 11;14(6):R55. doi: 10.1186/gb-2013-14-6-r55.
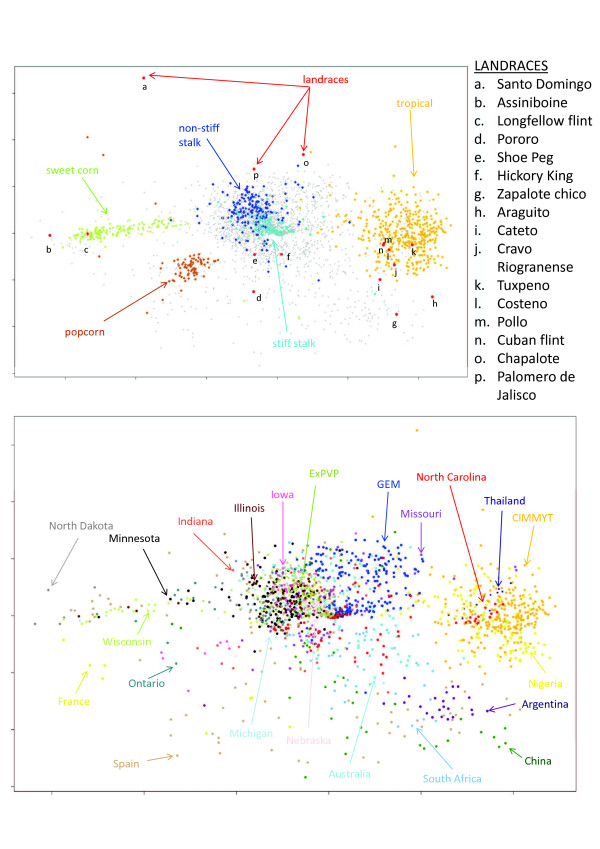
Genotyping by sequencing, a new low-cost, high-throughput sequencing technology was used to genotype 2,815 maize inbred accessions, preserved mostly at the National Plant Germplasm System in the USA. The collection includes inbred lines from breeding programs all over the world.
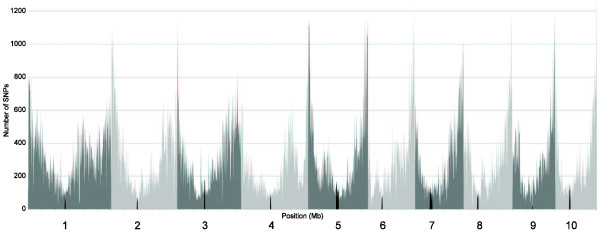
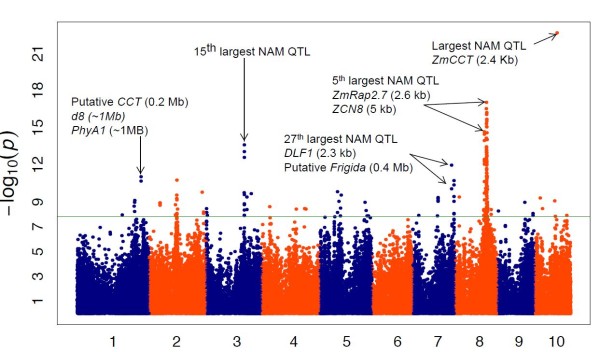
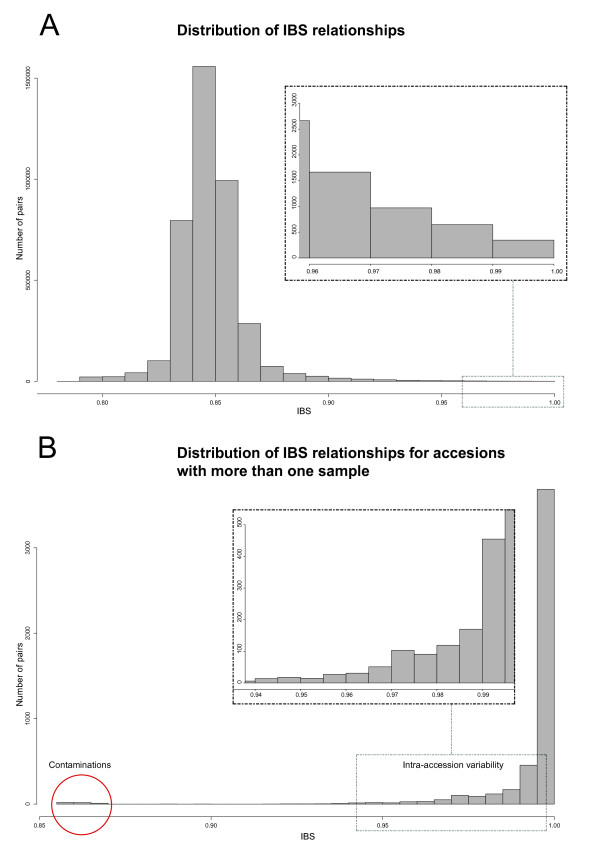
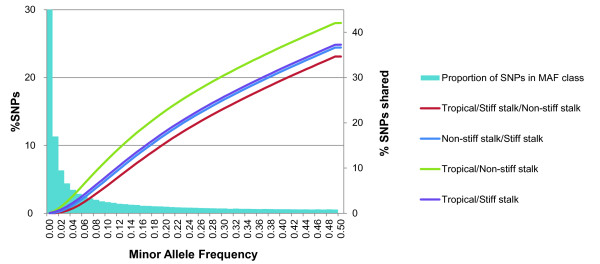
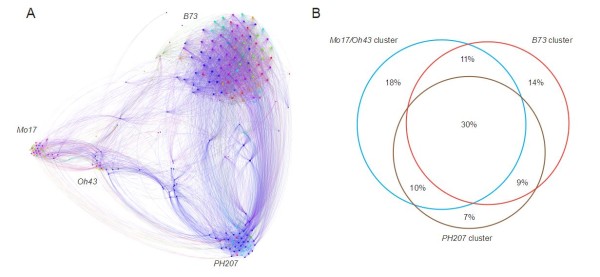
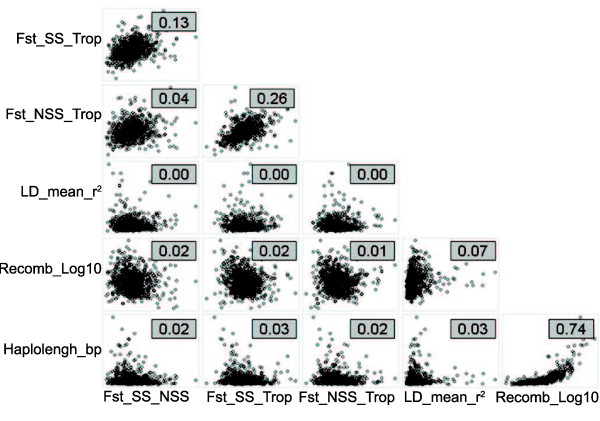
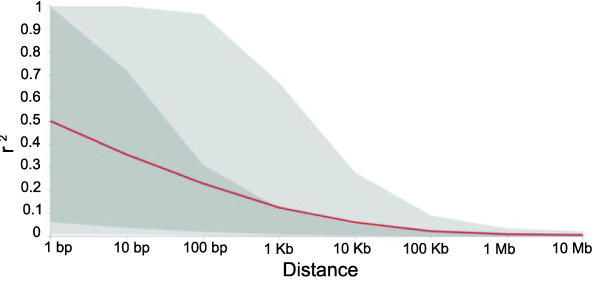
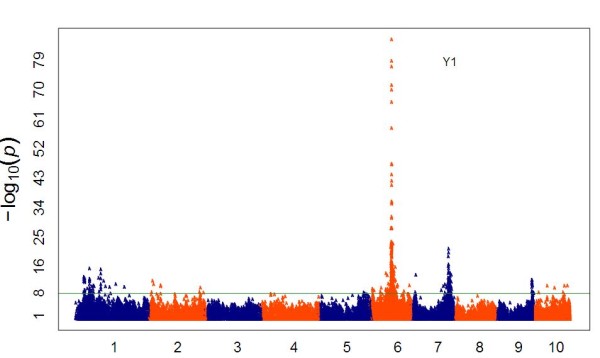
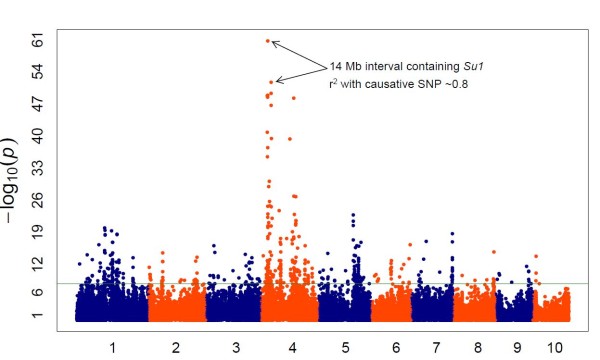
The method produced 681,257 single-nucleotide polymorphism (SNP) markers distributed across the entire genome, with the ability to detect rare alleles at high confidence levels. More than half of the SNPs in the collection are rare. Although most rare alleles have been incorporated into public temperate breeding programs, only a modest amount of the available diversity is present in the commercial germplasm. Analysis of genetic distances shows population stratification, including a small number of large clusters centered on key lines. Nevertheless, an average fixation index of 0.06 indicates moderate differentiation between the three major maize subpopulations. Linkage disequilibrium (LD) decays very rapidly, but the extent of LD is highly dependent on the particular group of germplasm and region of the genome. The utility of these data for performing genome-wide association studies was tested with two simply inherited traits and one complex trait. We identified trait associations at SNPs very close to known candidate genes for kernel color, sweet corn, and flowering time; however, results suggest that more SNPs are needed to better explore the genetic architecture of complex traits.
The genotypic information described here allows this publicly available panel to be exploited by researchers facing the challenges of sustainable agriculture through better knowledge of the nature of genetic diversity.
测序基因分型是一种新型低成本、高通量测序技术,用于对2815份玉米自交系种质进行基因分型,这些种质大多保存在美国国家植物种质系统中。该种质库包含来自世界各地育种项目的自交系。
该方法产生了681257个单核苷酸多态性(SNP)标记,分布于整个基因组,能够高置信度地检测稀有等位基因。该种质库中超过一半的SNP是稀有的。尽管大多数稀有等位基因已被纳入公共温带育种项目,但商业种质中仅存在少量可用的多样性。遗传距离分析显示了群体分层,包括以关键品系为中心的少数大簇。然而,平均固定指数为0.06表明三个主要玉米亚群之间存在中等程度的分化。连锁不平衡(LD)衰减非常迅速,但LD的程度高度依赖于特定的种质组和基因组区域。利用两个简单遗传性状和一个复杂性状对这些数据进行全基因组关联研究的效用进行了测试。我们在与已知的籽粒颜色、甜玉米和开花时间候选基因非常接近的SNP处鉴定出了性状关联;然而,结果表明需要更多的SNP来更好地探索复杂性状的遗传结构。
这里描述的基因型信息使这个公开可用的群体能够被研究人员利用,通过更好地了解遗传多样性的本质来应对可持续农业面临的挑战。