Department of Entomology, College of Natural Resources and Environment, South China Agricultural University, Guangzhou, PR China.
PLoS One. 2013 Jul 16;8(7):e69543. doi: 10.1371/journal.pone.0069543. Print 2013.
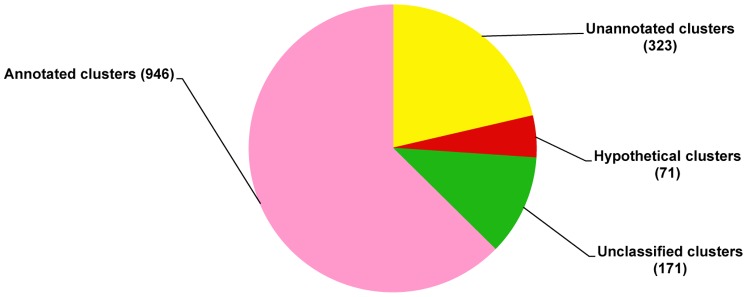
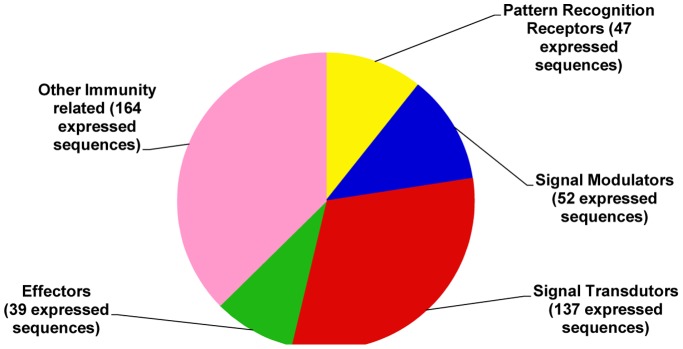
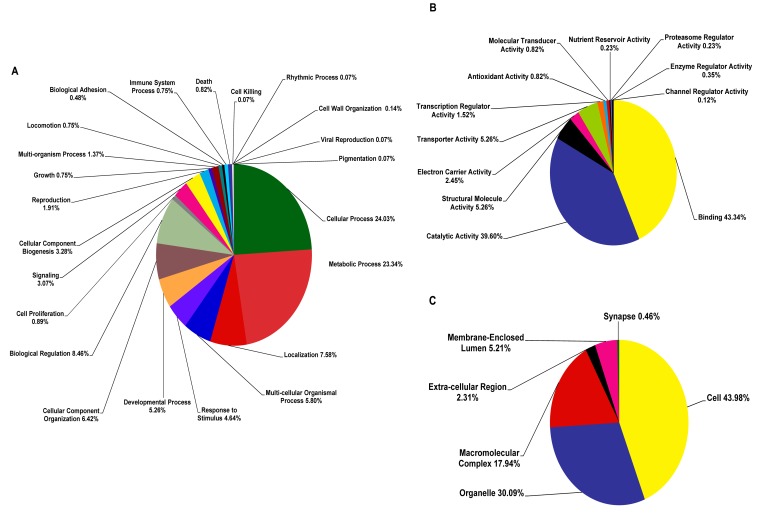
Formosan subterranean termites, Coptotermes formosanus Shiraki, live socially in microbial-rich habitats. To understand the molecular mechanism by which termites combat pathogenic microbes, a full-length normalized cDNA library and four Suppression Subtractive Hybridization (SSH) libraries were constructed from termite workers infected with entomopathogenic fungi (Metarhizium anisopliae and Beauveria bassiana), Gram-positive Bacillus thuringiensis and Gram-negative Escherichia coli, and the libraries were analyzed. From the high quality normalized cDNA library, 439 immune-related sequences were identified. These sequences were categorized as pattern recognition receptors (47 sequences), signal modulators (52 sequences), signal transducers (137 sequences), effectors (39 sequences) and others (164 sequences). From the SSH libraries, 27, 17, 22 and 15 immune-related genes were identified from each SSH library treated with M. anisopliae, B. bassiana, B. thuringiensis and E. coli, respectively. When the normalized cDNA library was compared with the SSH libraries, 37 immune-related clusters were found in common; 56 clusters were identified in the SSH libraries, and 259 were identified in the normalized cDNA library. The immune-related gene expression pattern was further investigated using quantitative real time PCR (qPCR). Important immune-related genes were characterized, and their potential functions were discussed based on the integrated analysis of the results. We suggest that normalized cDNA and SSH libraries enable us to discover functional genes transcriptome. The results remarkably expand our knowledge about immune-inducible genes in C. formosanus Shiraki and enable the future development of novel control strategies for the management of Formosan subterranean termites.
台湾乳白蚁,Coptotermes formosanus Shiraki,生活在微生物丰富的群居环境中。为了了解白蚁对抗病原微生物的分子机制,我们构建了一个全长标准化 cDNA 文库和四个抑制性消减杂交(SSH)文库,这些文库来自感染了昆虫病原真菌(绿僵菌和球孢白僵菌)、革兰氏阳性菌苏云金芽孢杆菌和革兰氏阴性菌大肠杆菌的工蚁。从高质量的标准化 cDNA 文库中,鉴定出了 439 个与免疫相关的序列。这些序列被分为模式识别受体(47 个序列)、信号调节剂(52 个序列)、信号转导器(137 个序列)、效应子(39 个序列)和其他(164 个序列)。从每个 SSH 文库中,分别鉴定出了来自绿僵菌、球孢白僵菌、苏云金芽孢杆菌和大肠杆菌处理的 SSH 文库中的 27、17、22 和 15 个与免疫相关的基因。当将标准化 cDNA 文库与 SSH 文库进行比较时,发现有 37 个免疫相关簇是共同的;在 SSH 文库中鉴定出 56 个簇,在标准化 cDNA 文库中鉴定出 259 个簇。进一步使用定量实时 PCR(qPCR)研究了免疫相关基因的表达模式。根据对结果的综合分析,对重要的免疫相关基因进行了特征描述,并讨论了它们的潜在功能。我们认为,标准化 cDNA 和 SSH 文库使我们能够发现功能基因转录组。这些结果显著扩展了我们对 C. formosanus Shiraki 中免疫诱导基因的认识,并为未来开发新型控制策略来管理台湾乳白蚁提供了依据。