School of Information and Control, Nanjing University of Information Science and Technology, Nanjing, Jiangsu, China.
PLoS One. 2013 Jul 25;8(7):e69842. doi: 10.1371/journal.pone.0069842. Print 2013.
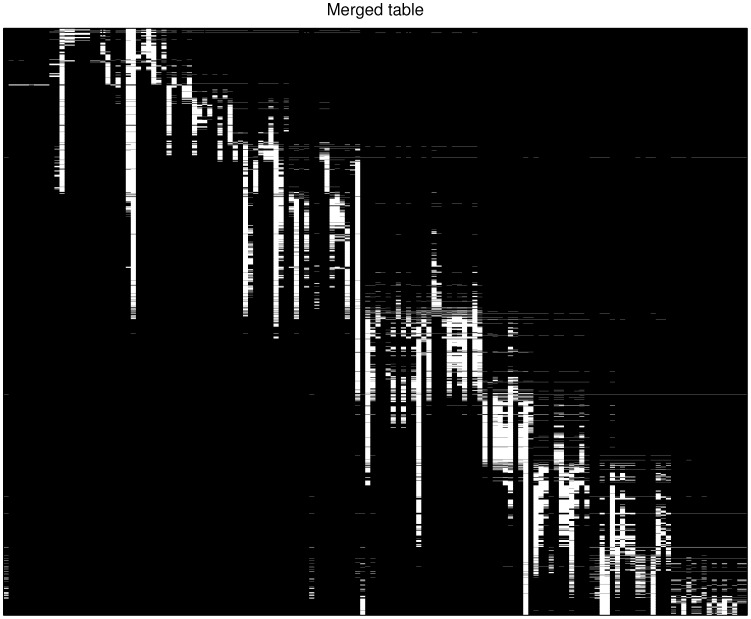
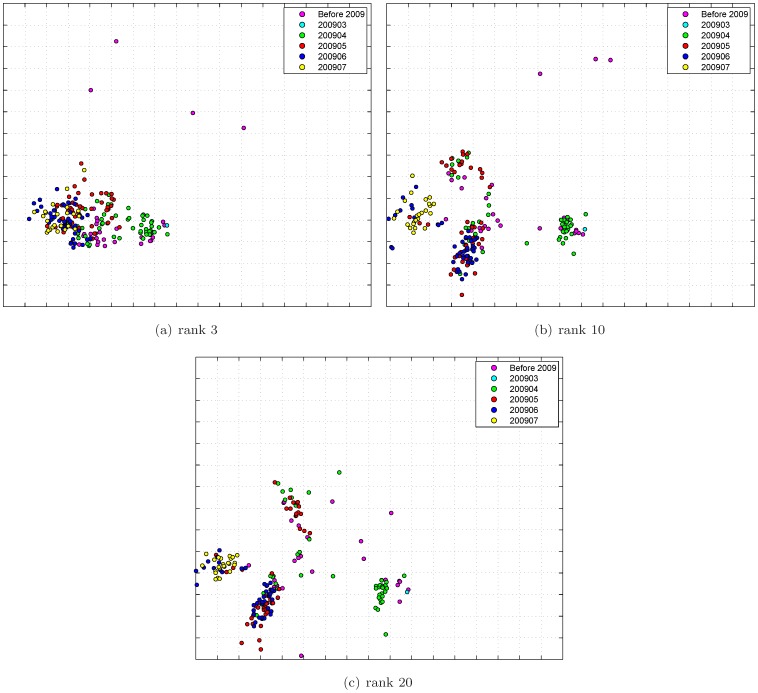
Antigenic characterization based on serological data, such as Hemagglutination Inhibition (HI) assay, is one of the routine procedures for influenza vaccine strain selection. In many cases, it would be impossible to measure all pairwise antigenic correlations between testing antigens and reference antisera in each individual experiment. Thus, we have to combine and integrate the HI tables from a number of individual experiments. Measurements from different experiments may be inconsistent due to different experimental conditions. Consequently we will observe a matrix with missing data and possibly inconsistent measurements. In this paper, we develop a new mathematical model, which we refer to as Joint Matrix Completion and Filtering, for HI data integration. In this approach, we simultaneously handle the incompleteness and uncertainty of observations by assuming that the underlying merged HI data matrix has low rank, as well as carefully modeling different levels of noises in each individual table. An efficient blockwise coordinate descent procedure is developed for optimization. The performance of our approach is validated on synthetic and real influenza datasets. The proposed joint matrix completion and filtering model can be adapted as a general model for biological data integration, targeting data noises and missing values within and across experiments.
基于血清学数据(如血凝抑制(HI)测定)的抗原特征分析是流感疫苗株选择的常规程序之一。在许多情况下,在每个单独的实验中,不可能测量测试抗原和参考抗血清之间的所有两两抗原相关性。因此,我们必须结合和整合来自许多单独实验的 HI 表。由于实验条件不同,来自不同实验的测量可能不一致。因此,我们将观察到一个具有缺失数据和可能不一致测量值的矩阵。在本文中,我们开发了一种新的数学模型,称为联合矩阵完成和滤波,用于 HI 数据集成。在这种方法中,我们通过假设基础合并 HI 数据矩阵具有低秩来同时处理观测值的不完整性和不确定性,并且仔细模拟每个单独表中的不同噪声水平。开发了一种有效的块坐标下降方法进行优化。我们的方法在合成和真实流感数据集上的性能验证。所提出的联合矩阵完成和滤波模型可以作为针对实验内和实验间数据噪声和缺失值的生物数据集成的通用模型进行适配。