Department of Basic Sciences, College of Veterinary Medicine, Mississippi State University, Mississippi State, Mississippi, USA.
PLoS Comput Biol. 2010 Oct 7;6(10):e1000949. doi: 10.1371/journal.pcbi.1000949.
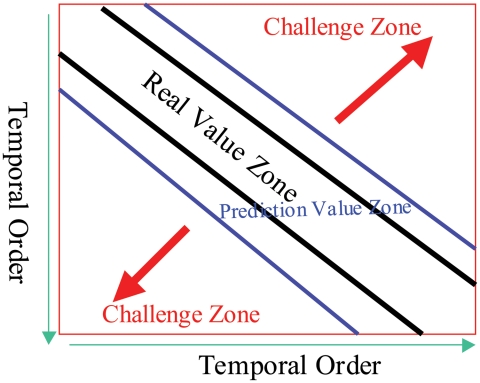
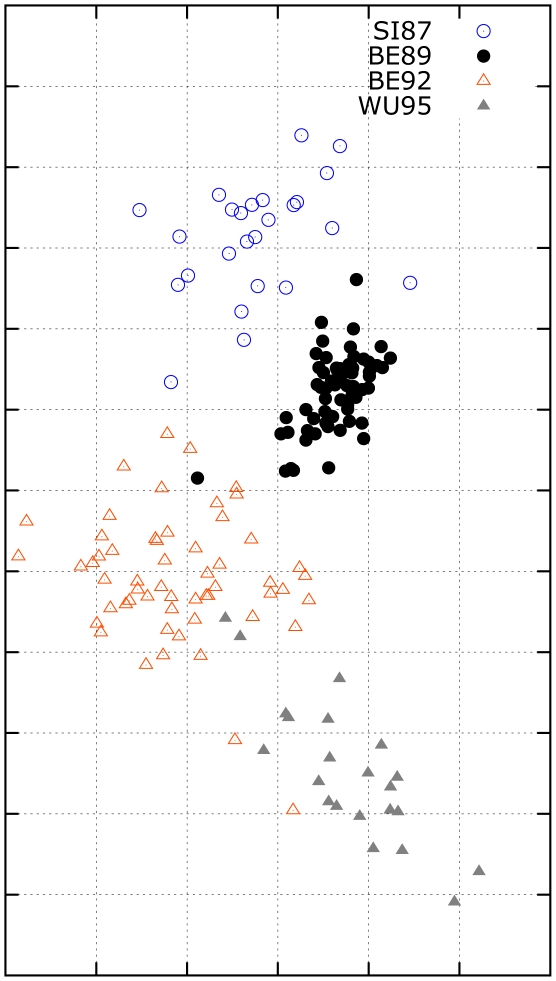
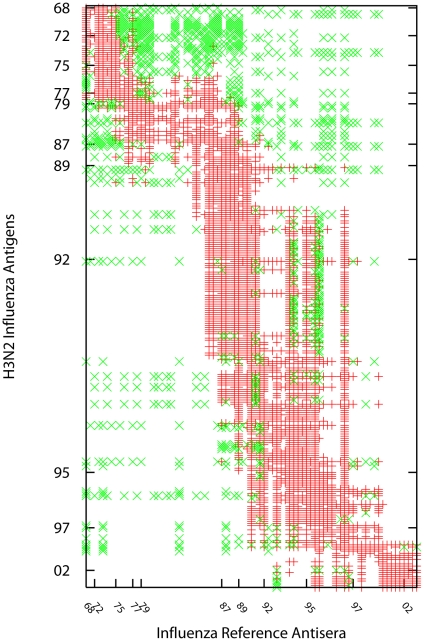
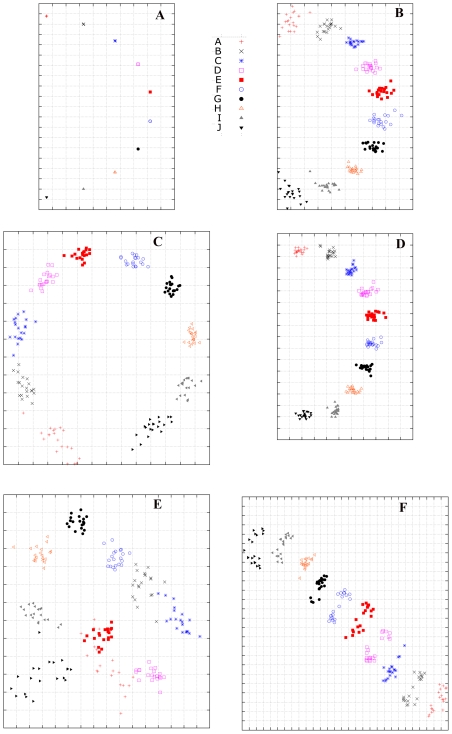
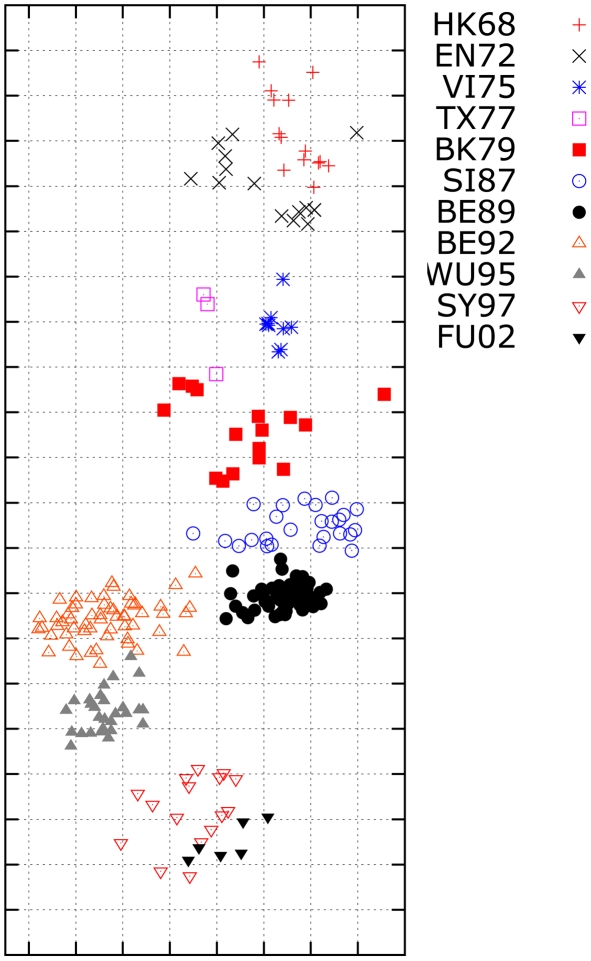
Influenza viruses have been responsible for large losses of lives around the world and continue to present a great public health challenge. Antigenic characterization based on hemagglutination inhibition (HI) assay is one of the routine procedures for influenza vaccine strain selection. However, HI assay is only a crude experiment reflecting the antigenic correlations among testing antigens (viruses) and reference antisera (antibodies). Moreover, antigenic characterization is usually based on more than one HI dataset. The combination of multiple datasets results in an incomplete HI matrix with many unobserved entries. This paper proposes a new computational framework for constructing an influenza antigenic cartography from this incomplete matrix, which we refer to as Matrix Completion-Multidimensional Scaling (MC-MDS). In this approach, we first reconstruct the HI matrices with viruses and antibodies using low-rank matrix completion, and then generate the two-dimensional antigenic cartography using multidimensional scaling. Moreover, for influenza HI tables with herd immunity effect (such as those from Human influenza viruses), we propose a temporal model to reduce the inherent temporal bias of HI tables caused by herd immunity. By applying our method in HI datasets containing H3N2 influenza A viruses isolated from 1968 to 2003, we identified eleven clusters of antigenic variants, representing all major antigenic drift events in these 36 years. Our results showed that both the completed HI matrix and the antigenic cartography obtained via MC-MDS are useful in identifying influenza antigenic variants and thus can be used to facilitate influenza vaccine strain selection. The webserver is available at http://sysbio.cvm.msstate.edu/AntigenMap.
流感病毒在全球范围内造成了巨大的生命损失,并且仍然是一个重大的公共卫生挑战。基于血凝抑制(HI)测定的抗原特征分析是流感疫苗株选择的常规程序之一。然而,HI 测定只是反映测试抗原(病毒)和参考抗血清(抗体)之间抗原相关性的一种粗略实验。此外,抗原特征分析通常基于多个 HI 数据集。多个数据集的组合导致 HI 矩阵不完整,存在许多未观察到的条目。本文提出了一种从这个不完整矩阵构建流感抗原图谱的新计算框架,我们称之为矩阵补全-多维尺度分析(MC-MDS)。在这种方法中,我们首先使用低秩矩阵补全重建病毒和抗体的 HI 矩阵,然后使用多维尺度分析生成二维抗原图谱。此外,对于具有群体免疫效应的 HI 表(如人类流感病毒的 HI 表),我们提出了一个时间模型来减少 HI 表中由群体免疫引起的固有时间偏差。通过在包含 1968 年至 2003 年分离的 H3N2 流感 A 病毒的 HI 数据集上应用我们的方法,我们确定了 11 个抗原变异体簇,代表了这 36 年来所有主要的抗原漂移事件。我们的结果表明,通过 MC-MDS 获得的完整 HI 矩阵和抗原图谱有助于识别流感抗原变异体,因此可用于促进流感疫苗株选择。该网络服务器可在 http://sysbio.cvm.msstate.edu/AntigenMap 上访问。