Szatkiewicz J P, Neale B M, O'Dushlaine C, Fromer M, Goldstein J I, Moran J L, Chambert K, Kähler A, Magnusson P K E, Hultman C M, Sklar P, Purcell S, McCarroll S A, Sullivan P F
Department of Genetics, University of North Carolina, Chapel Hill, NC, USA.
Mol Psychiatry. 2013 Nov;18(11):1178-84. doi: 10.1038/mp.2013.98. Epub 2013 Aug 13.
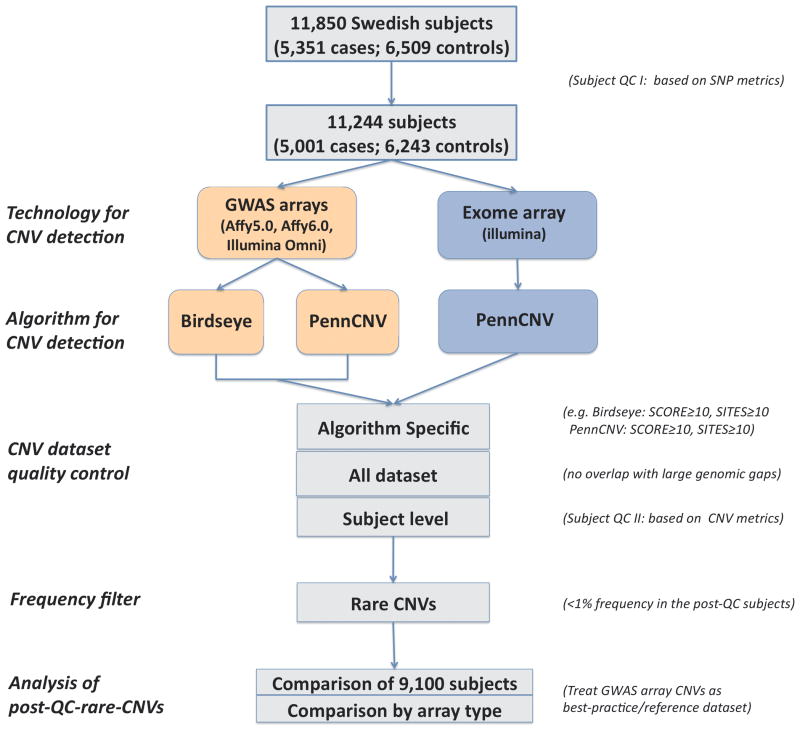
Although copy number variants (CNVs) are important in genomic medicine, CNVs have not been systematically assessed for many complex traits. Several large rare CNVs increase risk for schizophrenia (SCZ) and autism and often demonstrate pleiotropic effects; however, their frequencies in the general population and other complex traits are unknown. Genotyping large numbers of samples is essential for progress. Large cohorts from many different diseases are being genotyped using exome-focused arrays designed to detect uncommon or rare protein-altering sequence variation. Although these arrays were not designed for CNV detection, the hybridization intensity data generated in each experiment could, in principle, be used for gene-focused CNV analysis. Our goal was to evaluate the extent to which CNVs can be detected using data from one particular exome array (the Illumina Human Exome Bead Chip). We genotyped 9100 Swedish subjects (3962 cases with SCZ and 5138 controls) using both standard genome-wide association study (GWAS) and exome arrays. In comparison with CNVs detected using GWAS arrays, we observed high sensitivity and specificity for detecting genic CNVs 400 kb including known pathogenic CNVs along with replicating the literature finding that cases with SCZ had greater enrichment for genic CNVs. Our data confirm the association of SCZ with 16p11.2 duplications and 22q11.2 deletions, and suggest a novel association with deletions at 11q12.2. Our results suggest the utility of exome-focused arrays in surveying large genic CNVs in very large samples; and thereby open the door for new opportunities such as conducting well-powered CNV assessment and comparisons between different diseases. The use of a single platform also minimizes potential confounding factors that could impact accurate detection.
尽管拷贝数变异(CNV)在基因组医学中很重要,但尚未针对许多复杂性状对CNV进行系统评估。几种大型罕见CNV会增加精神分裂症(SCZ)和自闭症的风险,并且常常表现出多效性;然而,它们在普通人群中的频率以及与其他复杂性状的关系尚不清楚。对大量样本进行基因分型是取得进展的关键。来自许多不同疾病的大型队列正在使用旨在检测罕见或罕见蛋白质改变序列变异的外显子靶向阵列进行基因分型。尽管这些阵列并非设计用于检测CNV,但每个实验中产生的杂交强度数据原则上可用于基因靶向CNV分析。我们的目标是评估使用来自一种特定外显子阵列(Illumina人类外显子芯片)的数据能够检测CNV的程度。我们使用标准的全基因组关联研究(GWAS)和外显子阵列对9100名瑞典受试者(3962例SCZ患者和5138名对照)进行了基因分型。与使用GWAS阵列检测到的CNV相比,我们观察到在检测包括已知致病CNV在内的400 kb基因CNV时具有高灵敏度和特异性,同时重现了文献中的发现,即SCZ患者的基因CNV富集程度更高。我们的数据证实了SCZ与16p11.2重复和22q11.2缺失的关联,并提示与11q12.2缺失存在新的关联。我们的结果表明外显子靶向阵列在对非常大的样本中的大型基因CNV进行检测时具有实用性;从而为新的机会打开了大门,例如进行有充分统计学效力的CNV评估以及不同疾病之间的比较。使用单一平台还能最大程度减少可能影响准确检测的潜在混杂因素。