Tanz Centre for Research in Neurodegenerative Diseases, University of Toronto, Toronto, Ontario, Canada.
Taub Institute for Research on Alzheimer's Disease and the Aging Brain, Gertrude H. Sergievsky Center, Departments of Neurology, Psychiatry, and Medicine, College of Physicians and Surgeons, Columbia University, New York, New York3Department of Epidemiolo.
JAMA Neurol. 2013 Oct;70(10):1261-7. doi: 10.1001/jamaneurol.2013.3545.
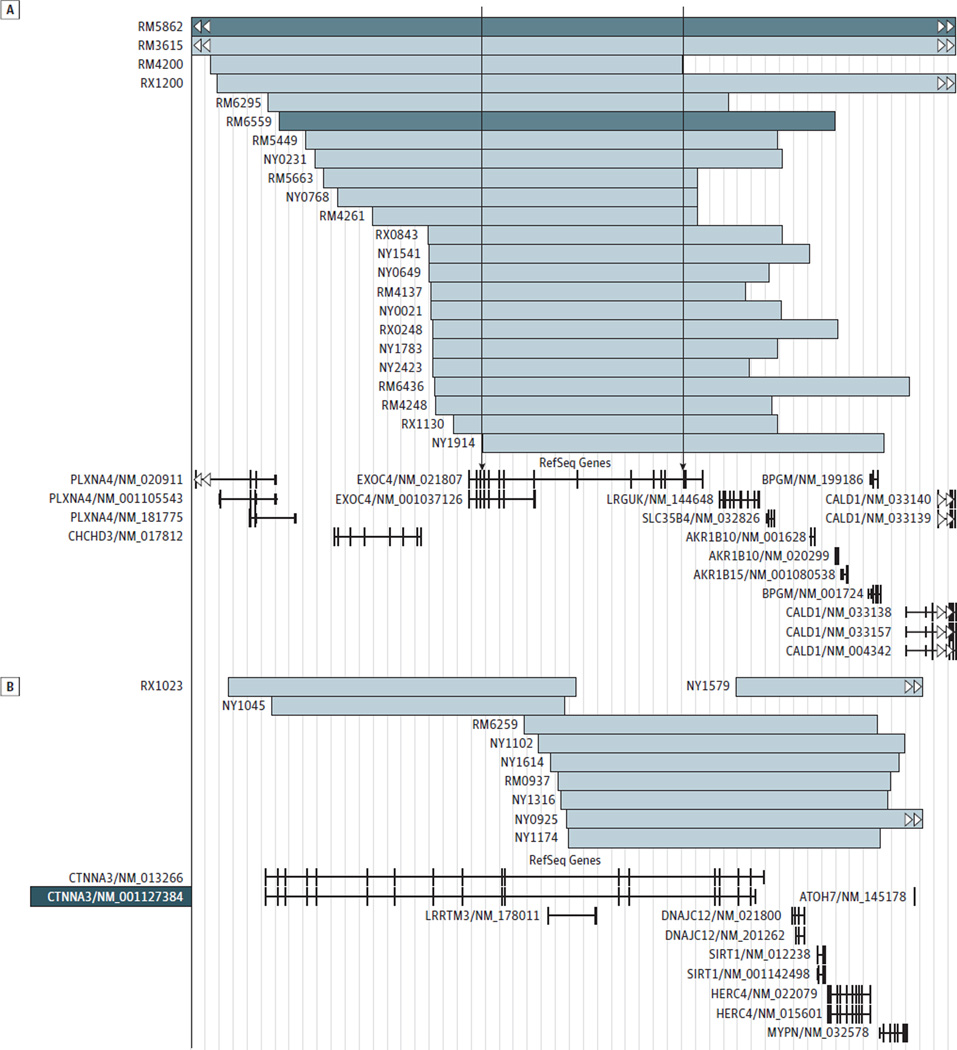
IMPORTANCE The search for novel Alzheimer disease (AD) genes or pathologic mutations within known AD loci is ongoing. The development of array technologies has helped to identify rare recessive mutations among long runs of homozygosity (ROHs), in which both parental alleles are identical. Caribbean Hispanics are known to have an elevated risk for AD and tend to have large families with evidence of inbreeding. OBJECTIVE To test the hypothesis that the late-onset AD in a Caribbean Hispanic population might be explained in part by the homozygosity of unknown loci that could harbor recessive AD risk haplotypes or pathologic mutations. DESIGN We used genome-wide array data to identify ROHs (>1 megabase) and conducted global burden and locus-specific ROH analyses. SETTING A whole-genome case-control ROH study. PARTICIPANTS A Caribbean Hispanic data set of 547 unrelated cases (48.8% with familial AD) and 542 controls collected from a population known to have a 3-fold higher risk of AD vs non-Hispanics in the same community. Based on a Structure program analysis, our data set consisted of African Hispanic (207 cases and 192 controls) and European Hispanic (329 cases and 326 controls) participants. EXPOSURE Alzheimer disease risk genes. MAIN OUTCOMES AND MEASURES We calculated the total and mean lengths of the ROHs per sample. Global burden measurements among autosomal chromosomes were investigated in cases vs controls. Pools of overlapping ROH segments (consensus regions) were identified, and the case to control ratio was calculated for each consensus region. We formulated the tested hypothesis before data collection. RESULTS In total, we identified 17 137 autosomal regions with ROHs. The mean length of the ROH per person was significantly greater in cases vs controls (P = .0039), and this association was stronger with familial AD (P = .0005). Among the European Hispanics, a consensus region at the EXOC4 locus was significantly associated with AD even after correction for multiple testing (empirical P value 1 [EMP1], .0001; EMP2, .002; 21 AD cases vs 2 controls). Among the African Hispanic subset, the most significant but nominal association was observed for CTNNA3, a well-known AD gene candidate (EMP1, .002; 10 AD cases vs 0 controls). CONCLUSIONS AND RELEVANCE Our results show that ROHs could significantly contribute to the etiology of AD. Future studies would require the analysis of larger, relatively inbred data sets that might reveal novel recessive AD genes. The next step is to conduct sequencing of top significant loci in a subset of samples with overlapping ROHs.
目前仍在寻找新的阿尔茨海默病(AD)基因或已知 AD 基因座内的病理突变。阵列技术的发展有助于识别长串联同源性(ROH)中的罕见隐性突变,其中父母双方的等位基因是相同的。众所周知,加勒比西班牙裔人群患 AD 的风险较高,且往往有大型家庭,存在近亲繁殖的证据。目的:检验假设,即加勒比西班牙裔人群的迟发性 AD 部分可能是由未知基因座的纯合性解释的,这些基因座可能携带隐性 AD 风险单体型或病理突变。设计:我们使用全基因组阵列数据来识别 ROH(>1 兆碱基),并进行了全基因组负担和基因座特异性 ROH 分析。地点:全基因组病例对照 ROH 研究。参与者:来自一个社区的 547 名无血缘关系的病例(48.8%有家族性 AD)和 542 名对照,这些病例来自一个已知 AD 风险比非西班牙裔人群高 3 倍的人群。基于 Structure 程序分析,我们的数据集中包括非洲西班牙裔(207 例和 192 例对照)和欧洲西班牙裔(329 例和 326 例对照)参与者。暴露:AD 风险基因。主要结果和措施:我们计算了每个样本的 ROH 总长度和平均长度。在病例与对照之间研究了常染色体上的全基因组负担测量。确定了重叠 ROH 片段(共识区域)的池,并计算了每个共识区域的病例与对照比值。在数据收集之前,我们提出了经过测试的假设。结果:总共确定了 17137 个具有 ROH 的常染色体区域。与对照组相比,病例组的 ROH 人均长度显著增加(P=0.0039),并且这种关联在家族性 AD 中更强(P=0.0005)。在欧洲西班牙裔人群中,EXOC4 基因座的一个共识区域与 AD 显著相关,即使在经过多次检验校正后也是如此(经验 P 值 1[EMP1],.0001;EMP2,.002;21 例 AD 病例与 2 例对照)。在非洲西班牙裔亚组中,CTNNA3 观察到最显著但名义上的关联,CTNNA3 是一个众所周知的 AD 基因候选物(EMP1,.002;10 例 AD 病例与 0 例对照)。结论和相关性:我们的研究结果表明,ROH 可能对 AD 的发病机制有显著贡献。未来的研究需要分析更大的、相对近亲繁殖的数据集,这可能会揭示新的隐性 AD 基因。下一步是在具有重叠 ROH 的样本亚集中对顶级显著基因座进行测序。