Dept of Computer Science and Applied Mathematics, Weizmann Institute of Science, Rehovot, Israel.
PLoS Comput Biol. 2013;9(8):e1003200. doi: 10.1371/journal.pcbi.1003200. Epub 2013 Aug 22.
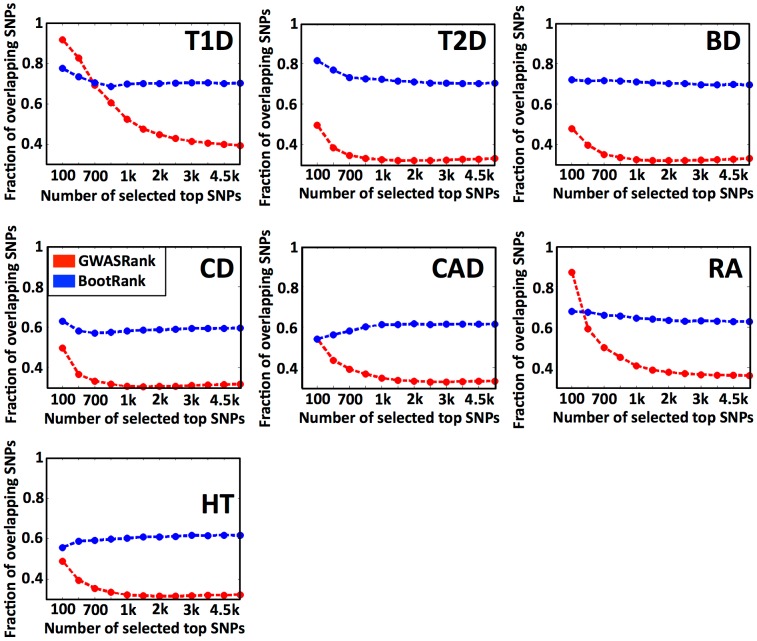
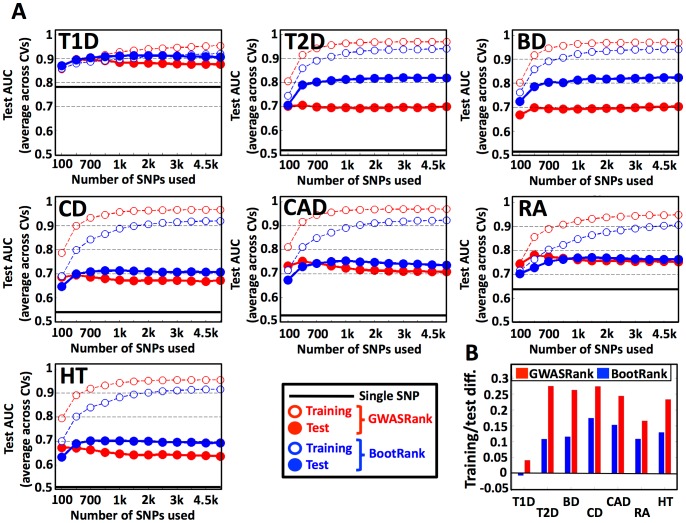
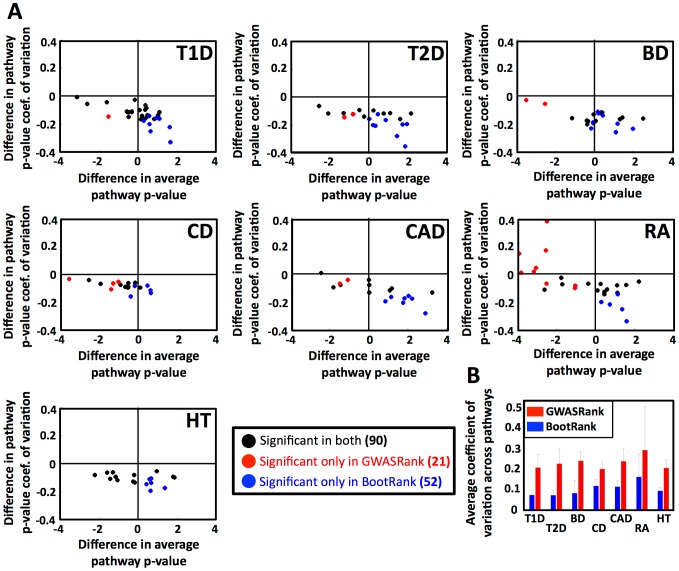
Genome-wide association studies (GWAS) are widely used to search for genetic loci that underlie human disease. Another goal is to predict disease risk for different individuals given their genetic sequence. Such predictions could either be used as a "black box" in order to promote changes in life-style and screening for early diagnosis, or as a model that can be studied to better understand the mechanism of the disease. Current methods for risk prediction typically rank single nucleotide polymorphisms (SNPs) by the p-value of their association with the disease, and use the top-associated SNPs as input to a classification algorithm. However, the predictive power of such methods is relatively poor. To improve the predictive power, we devised BootRank, which uses bootstrapping in order to obtain a robust prioritization of SNPs for use in predictive models. We show that BootRank improves the ability to predict disease risk of unseen individuals in the Wellcome Trust Case Control Consortium (WTCCC) data and results in a more robust set of SNPs and a larger number of enriched pathways being associated with the different diseases. Finally, we show that combining BootRank with seven different classification algorithms improves performance compared to previous studies that used the WTCCC data. Notably, diseases for which BootRank results in the largest improvements were recently shown to have more heritability than previously thought, likely due to contributions from variants with low minimum allele frequency (MAF), suggesting that BootRank can be beneficial in cases where SNPs affecting the disease are poorly tagged or have low MAF. Overall, our results show that improving disease risk prediction from genotypic information may be a tangible goal, with potential implications for personalized disease screening and treatment.
全基因组关联研究(GWAS)被广泛用于寻找人类疾病的遗传基因座。另一个目标是根据个体的遗传序列预测不同个体的疾病风险。这些预测可以作为“黑箱”,用于促进生活方式的改变和早期诊断筛查,也可以作为模型进行研究,以更好地了解疾病的机制。目前的风险预测方法通常根据与疾病关联的 p 值对单核苷酸多态性(SNP)进行排名,并将最相关的 SNP 用作分类算法的输入。然而,这些方法的预测能力相对较差。为了提高预测能力,我们设计了 BootRank,它使用了自举技术来获得 SNP 的稳健优先级,以便在预测模型中使用。我们表明,BootRank 提高了对未见个体疾病风险的预测能力,在惠康信托基金会病例对照研究(WTCCC)数据中,BootRank 导致与不同疾病相关的 SNP 更为稳健,富集途径数量更多。最后,我们表明,与之前使用 WTCCC 数据的研究相比,将 BootRank 与七种不同的分类算法相结合可以提高性能。值得注意的是,BootRank 结果导致改进最大的疾病最近被证明比以前认为的具有更高的遗传性,这可能是由于具有低最小等位基因频率(MAF)的变异体的贡献所致,这表明 BootRank 对于影响疾病的 SNP 标记不良或 MAF 较低的情况可能是有益的。总体而言,我们的研究结果表明,从基因型信息中提高疾病风险预测可能是一个切实可行的目标,这可能对个性化疾病筛查和治疗产生影响。