Department of Physical Chemistry, Fritz-Haber-Institut der Max-Planck-Gesellschaft, Berlin, Germany.
PLoS Comput Biol. 2013;9(8):e1003209. doi: 10.1371/journal.pcbi.1003209. Epub 2013 Aug 29.
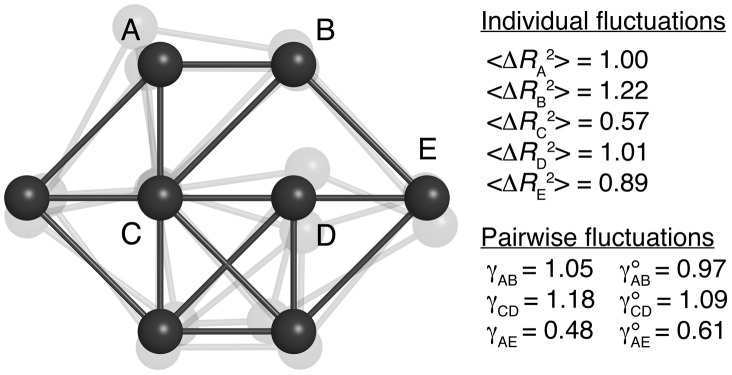
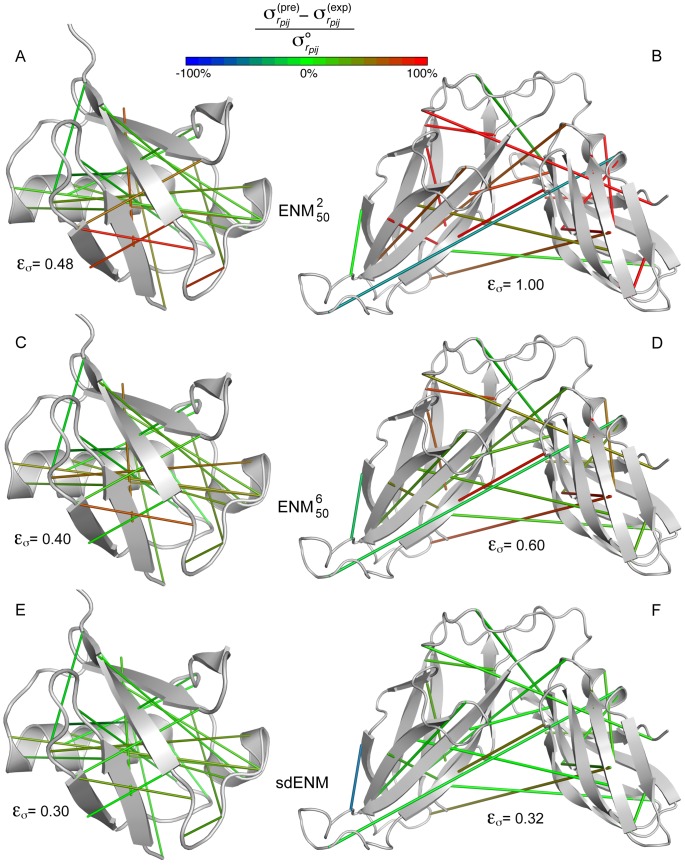
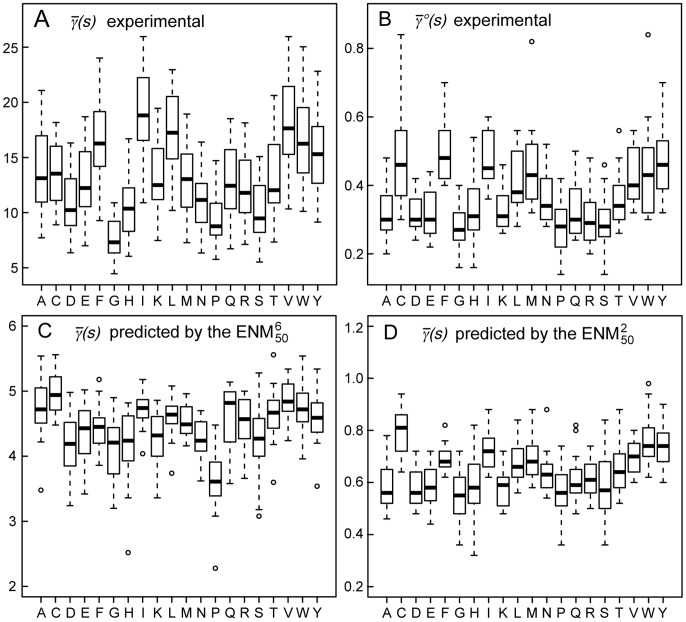
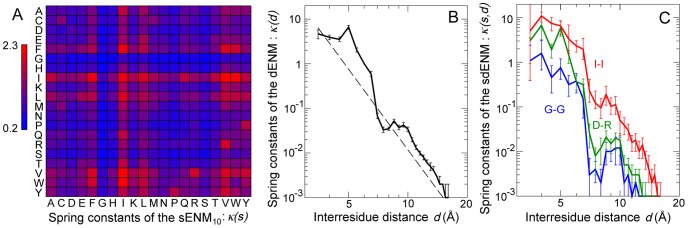
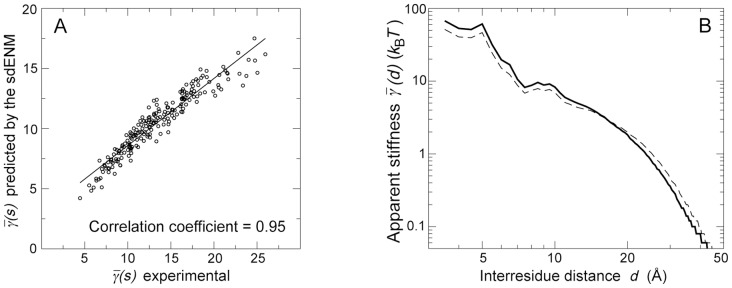
The proper biological functioning of proteins often relies on the occurrence of coordinated fluctuations around their native structure, or on their ability to perform wider and sometimes highly elaborated motions. Hence, there is considerable interest in the definition of accurate coarse-grained descriptions of protein dynamics, as an alternative to more computationally expensive approaches. In particular, the elastic network model, in which residue motions are subjected to pairwise harmonic potentials, is known to capture essential aspects of conformational dynamics in proteins, but has so far remained mostly phenomenological, and unable to account for the chemical specificities of amino acids. We propose, for the first time, a method to derive residue- and distance-specific effective harmonic potentials from the statistical analysis of an extensive dataset of NMR conformational ensembles. These potentials constitute dynamical counterparts to the mean-force statistical potentials commonly used for static analyses of protein structures. In the context of the elastic network model, they yield a strongly improved description of the cooperative aspects of residue motions, and give the opportunity to systematically explore the influence of sequence details on protein dynamics.
蛋白质的正常生物学功能通常依赖于其天然结构周围协调波动的发生,或者依赖于其执行更广泛、有时高度复杂运动的能力。因此,人们对定义准确的蛋白质动力学粗粒描述非常感兴趣,这是一种替代更昂贵的计算方法。特别是弹性网络模型,其中残基运动受到成对的谐波势的限制,已知可以捕获蛋白质构象动力学的基本方面,但到目前为止,它仍然主要是唯象的,并且无法解释氨基酸的化学特异性。我们首次提出了一种从大量 NMR 构象集合的统计分析中推导出残基和距离特异性有效谐波势的方法。这些势构成了与常用的蛋白质结构静态分析的平均力统计势的动力学对应物。在弹性网络模型的背景下,它们可以对残基运动的协同方面进行强有力的改进描述,并为系统地探索序列细节对蛋白质动力学的影响提供机会。