Department of Chemical Engineering, Indian Institute of Technology Bombay, Powai, Mumbai, India.
PLoS One. 2013 Aug 28;8(8):e71248. doi: 10.1371/journal.pone.0071248. eCollection 2013.
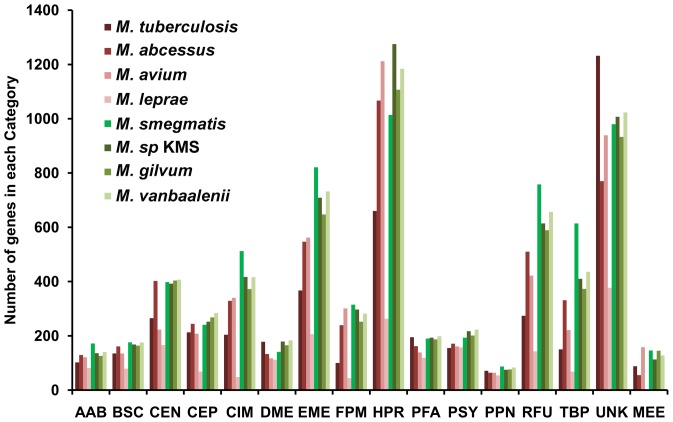
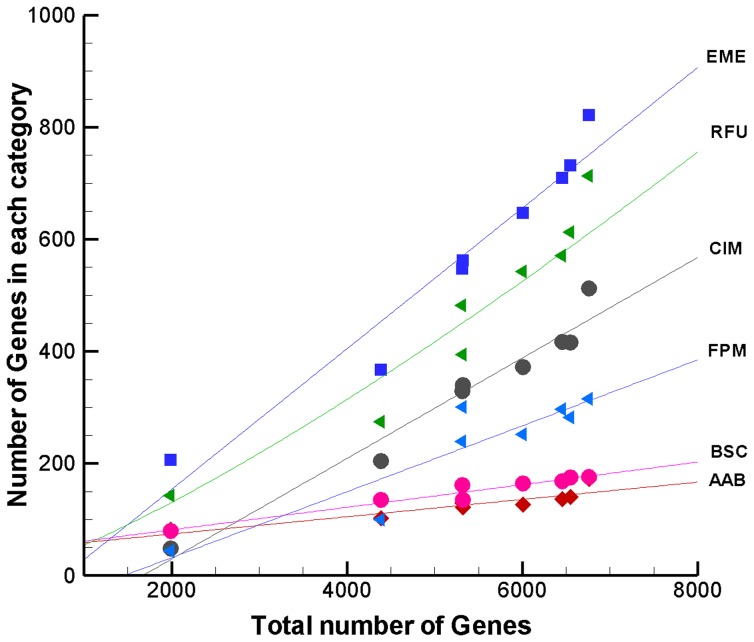
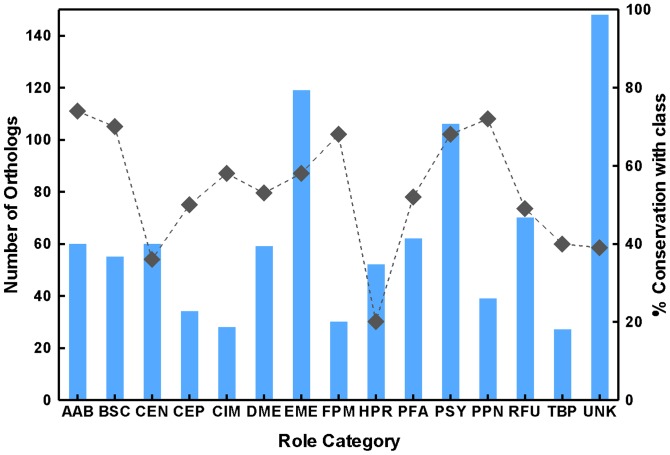
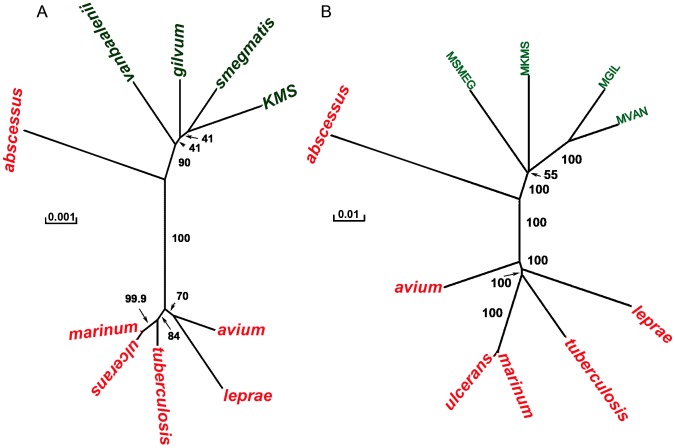
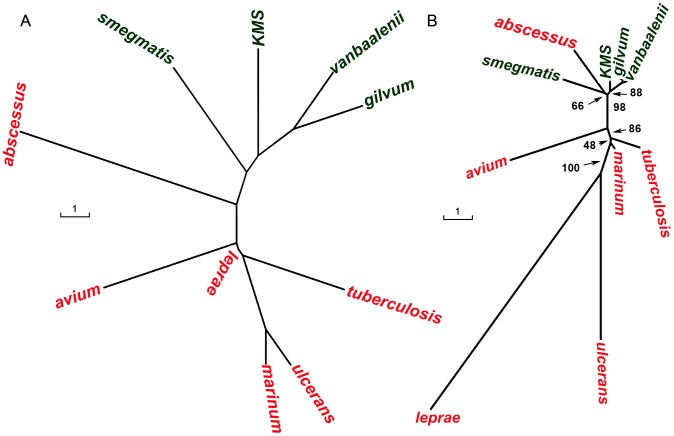
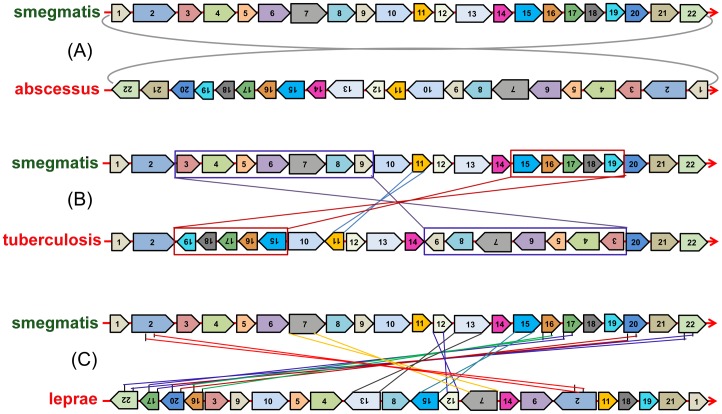
Mycobacterium species are the source of a variety of infectious diseases in a range of hosts. Genome based methods are used to understand the adaptation of each pathogenic species to its unique niche. In this work, we report the comparison of pathogenic and non-pathogenic Mycobacterium genomes. Phylogenetic trees were constructed using sequence of core orthologs, gene content and gene order. It is found that the genome based methods can better resolve the inter-species evolutionary distances compared to the conventional 16S based tree. Phylogeny based on gene order highlights distinct evolutionary characteristics as compared to the methods based on sequence, as illustrated by the shift in the relative position of M. abscessus. This difference in gene order among the Mycobacterium species is further investigated using a detailed synteny analysis. It is found that while rearrangements between some Mycobacterium genomes are local within synteny blocks, few possess global rearrangements across the genomes. The study illustrates how a combination of different genome based methods is essential to build a robust phylogenetic relationship between closely related organisms.
分枝杆菌属的物种是多种宿主感染性疾病的来源。基于基因组的方法被用于了解每种病原物种对其独特小生境的适应性。在这项工作中,我们报告了致病性和非致病性分枝杆菌基因组的比较。使用核心直系同源物的序列、基因内容和基因顺序构建了系统发育树。结果发现,与传统的基于 16S 的树相比,基于基因组的方法可以更好地解决种间进化距离。基于基因顺序的系统发育与基于序列的方法相比突出了不同的进化特征,如图中脓肿分枝杆菌相对位置的变化所示。通过详细的同线性分析进一步研究了分枝杆菌属物种之间的这种基因顺序差异。结果发现,虽然一些分枝杆菌基因组之间的重排在基因顺序块内是局部的,但很少有跨基因组的全局重排。该研究说明了如何结合不同的基于基因组的方法对于构建密切相关的生物体之间稳健的系统发育关系至关重要。