Department of Systems Biology, Technical University of Denmark, Lyngby, Denmark.
PLoS Genet. 2013;9(9):e1003741. doi: 10.1371/journal.pgen.1003741. Epub 2013 Sep 5.
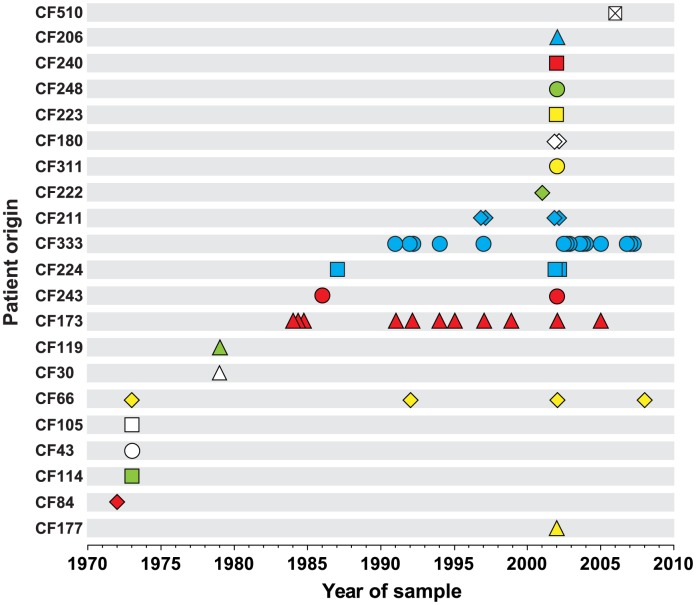
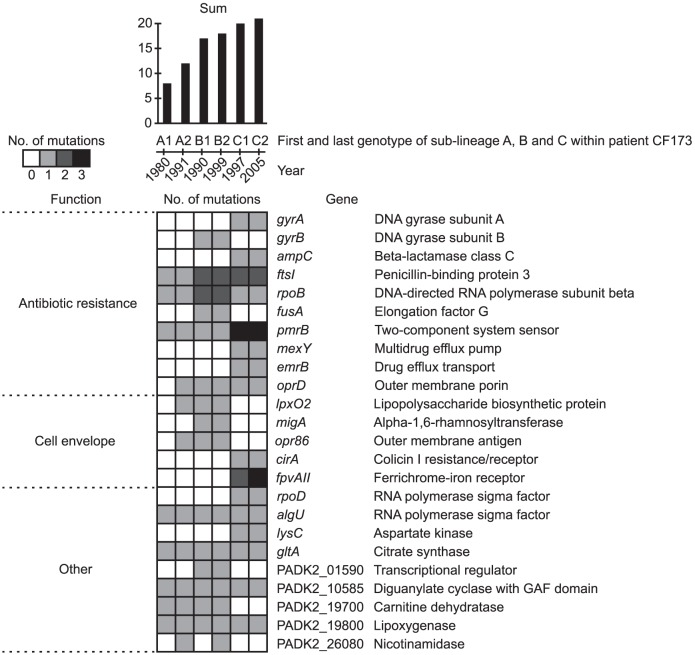
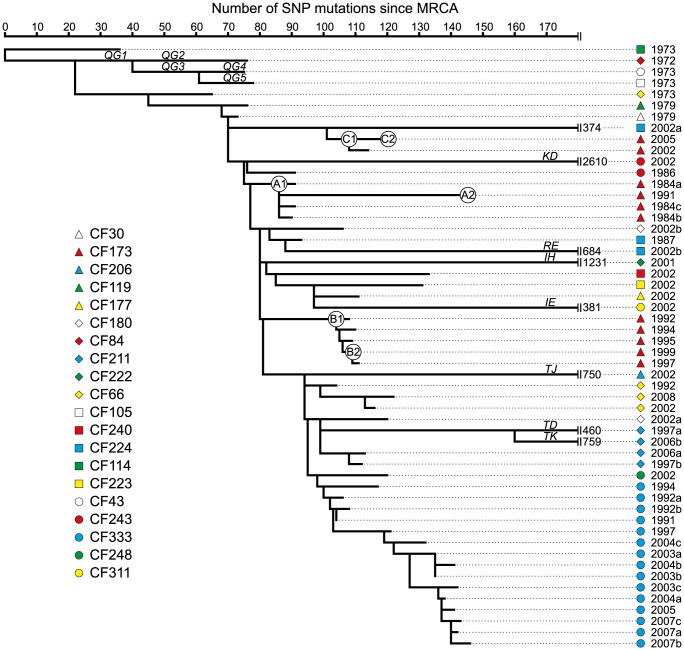
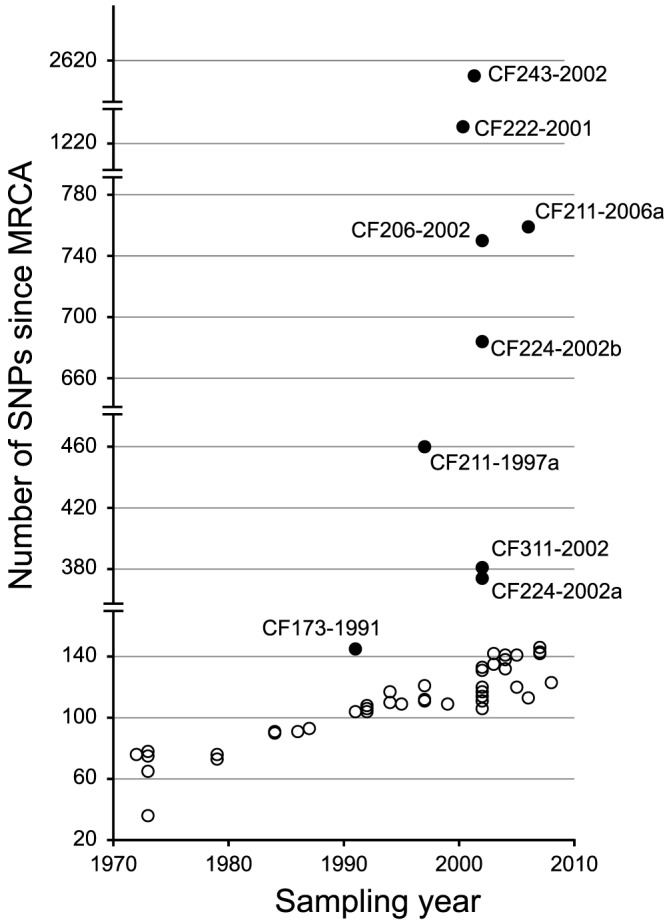
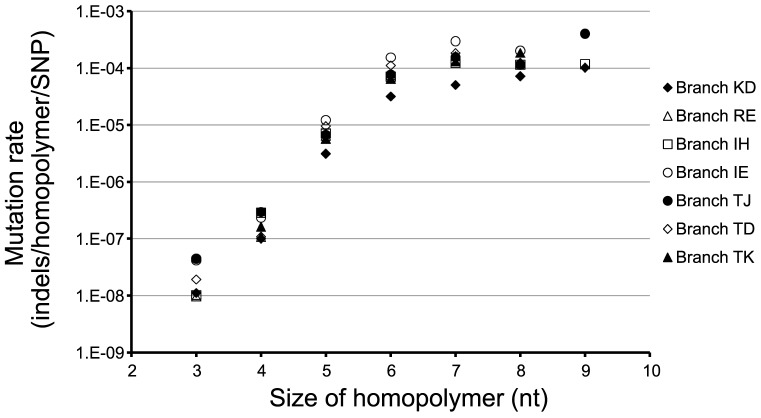
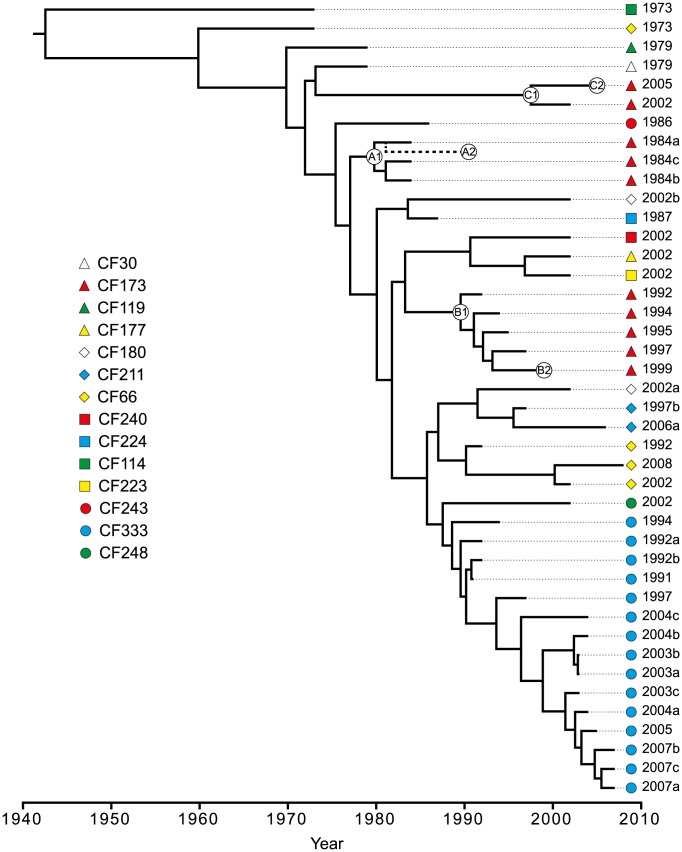
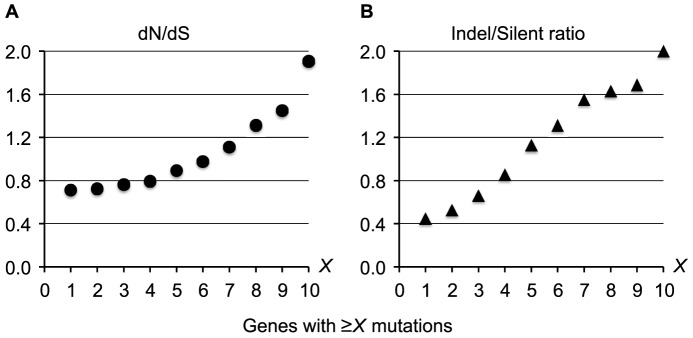
Genome sequencing of bacterial pathogens has advanced our understanding of their evolution, epidemiology, and response to antibiotic therapy. However, we still have only a limited knowledge of the molecular changes in in vivo evolving bacterial populations in relation to long-term, chronic infections. For example, it remains unclear what genes are mutated to facilitate the establishment of long-term existence in the human host environment, and in which way acquisition of a hypermutator phenotype with enhanced rates of spontaneous mutations influences the evolutionary trajectory of the pathogen. Here we perform a retrospective study of the DK2 clone type of P. aeruginosa isolated from Danish patients suffering from cystic fibrosis (CF), and analyze the genomes of 55 bacterial isolates collected from 21 infected individuals over 38 years. Our phylogenetic analysis of 8,530 mutations in the DK2 genomes shows that the ancestral DK2 clone type spread among CF patients through several independent transmission events. Subsequent to transmission, sub-lineages evolved independently for years in separate hosts, creating a unique possibility to study parallel evolution and identification of genes targeted by mutations to optimize pathogen fitness (pathoadaptive mutations). These genes were related to antibiotic resistance, the cell envelope, or regulatory functions, and we find that the prevalence of pathoadaptive mutations correlates with evolutionary success of co-evolving sub-lineages. The long-term co-existence of both normal and hypermutator populations enabled comparative investigations of the mutation dynamics in homopolymeric sequences in which hypermutators are particularly prone to mutations. We find a positive exponential correlation between the length of the homopolymer and its likelihood to acquire mutations and identify two homopolymer-containing genes preferentially mutated in hypermutators. This homopolymer facilitated differential mutagenesis provides a novel genome-wide perspective on the different evolutionary trajectories of hypermutators, which may help explain their emergence in CF infections.
细菌病原体的基因组测序提高了我们对其进化、流行病学和对抗生素治疗反应的认识。然而,我们对于与长期慢性感染相关的体内进化细菌群体的分子变化仍然知之甚少。例如,尚不清楚哪些基因发生突变以促进在人类宿主环境中的长期存在,以及获得具有更高自发突变率的超突变表型如何影响病原体的进化轨迹。在这里,我们对从丹麦囊性纤维化(CF)患者中分离出的铜绿假单胞菌 DK2 克隆型进行了回顾性研究,并分析了从 21 名受感染者中收集的 55 个细菌分离株的基因组。我们对 DK2 基因组中 8530 个突变的系统发育分析表明,祖先型 DK2 克隆型通过几个独立的传播事件在 CF 患者中传播。随后在传播后,亚谱系在不同的宿主中独立进化了数年,为研究平行进化和识别突变优化病原体适应性的目标基因(病理适应突变)创造了独特的可能性。这些基因与抗生素耐药性、细胞包膜或调节功能有关,我们发现病理适应突变的流行与共同进化的亚谱系的进化成功相关。正常和超突变体种群的长期共存使我们能够对同源多聚体序列中的突变动态进行比较研究,超突变体在同源多聚体序列中特别容易发生突变。我们发现,在同源多聚体的长度与其发生突变的可能性之间存在正指数相关性,并确定了两个在超突变体中优先突变的含有同源多聚体的基因。这种同源多聚体介导的差异诱变提供了一个全新的全基因组视角,了解超突变体的不同进化轨迹,这可能有助于解释它们在 CF 感染中的出现。