Department of Neurology and Neurotherapeutics, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA.
Department of Neurology and Neurotherapeutics, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA; Department of Psychiatry, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA.
Neurobiol Dis. 2014 Feb;62:113-23. doi: 10.1016/j.nbd.2013.09.009. Epub 2013 Sep 26.
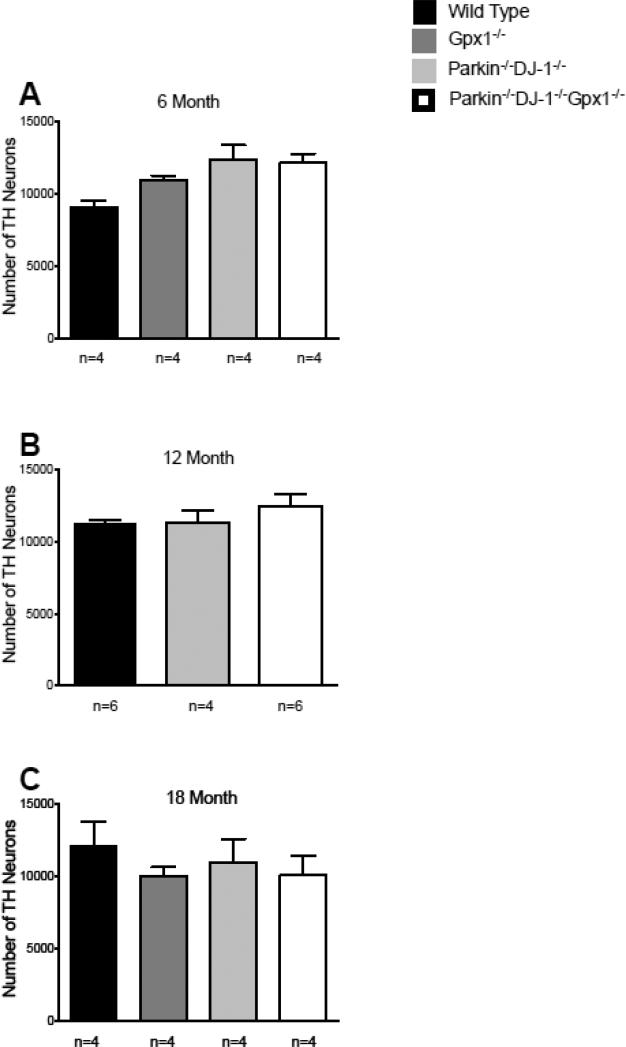
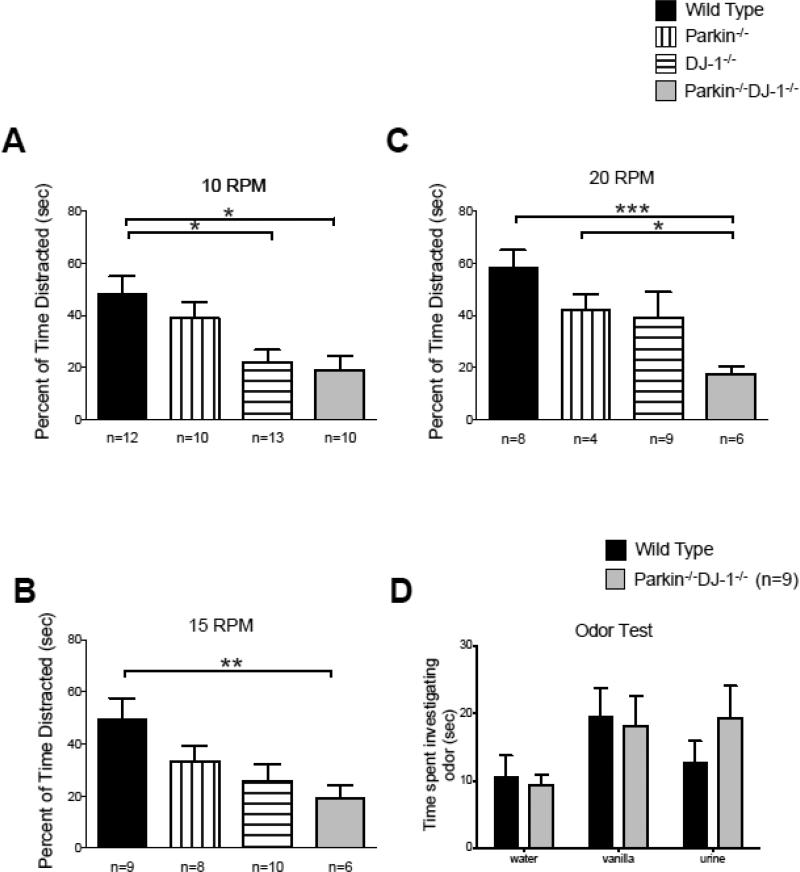
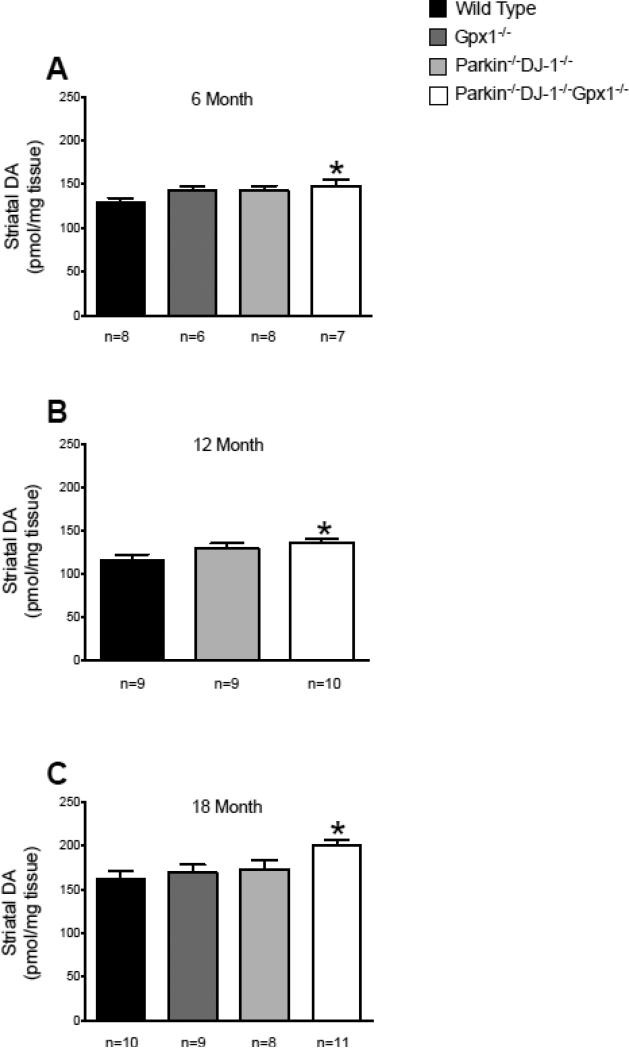
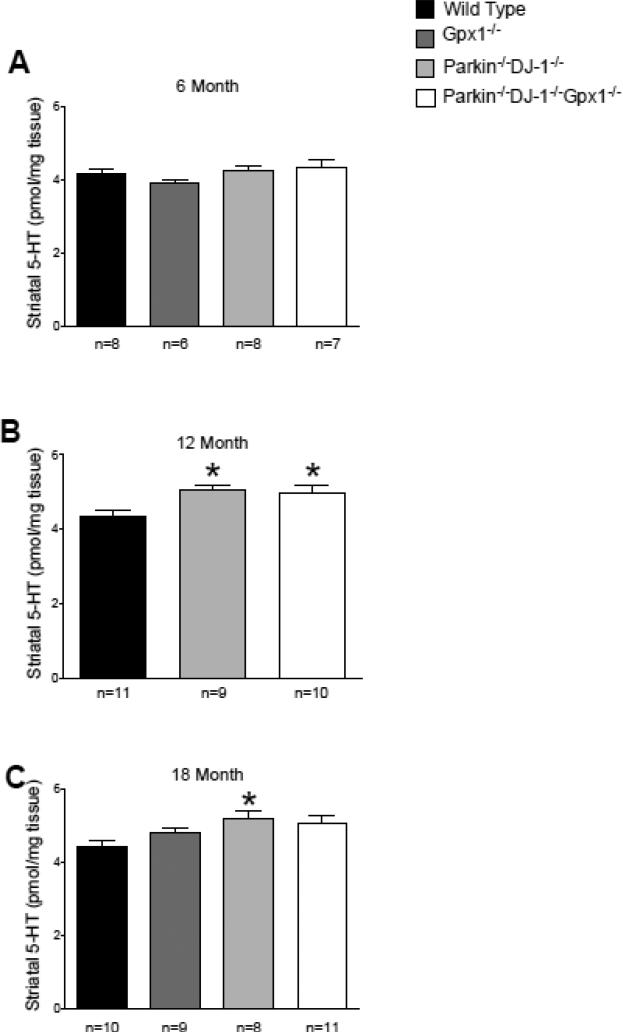
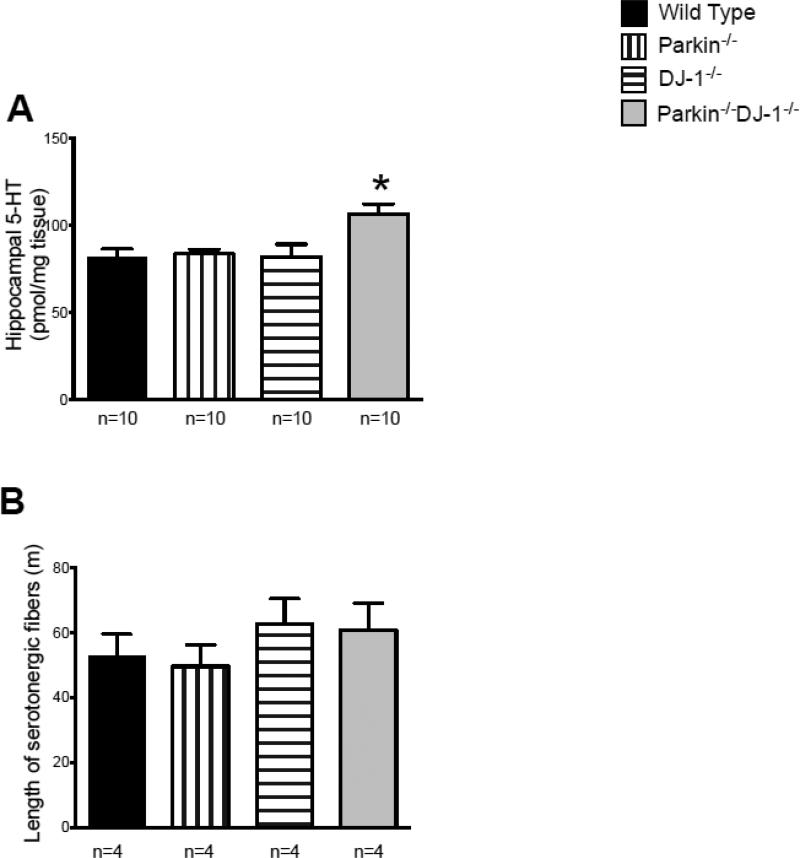
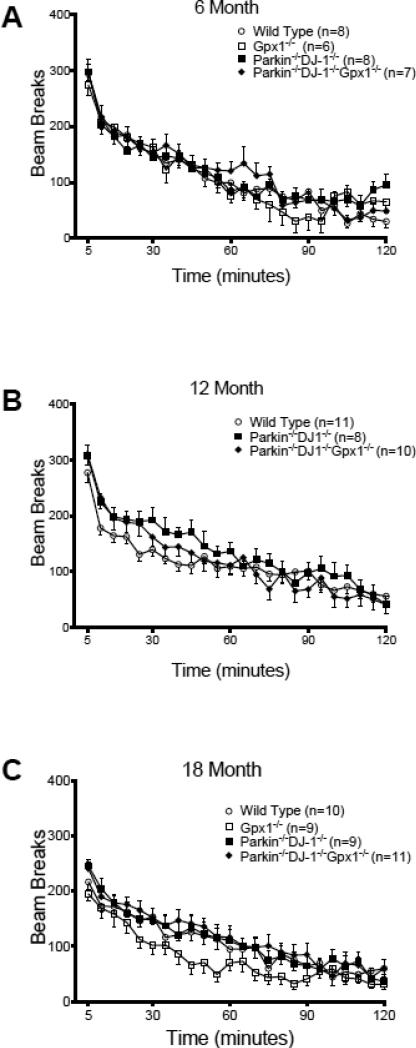
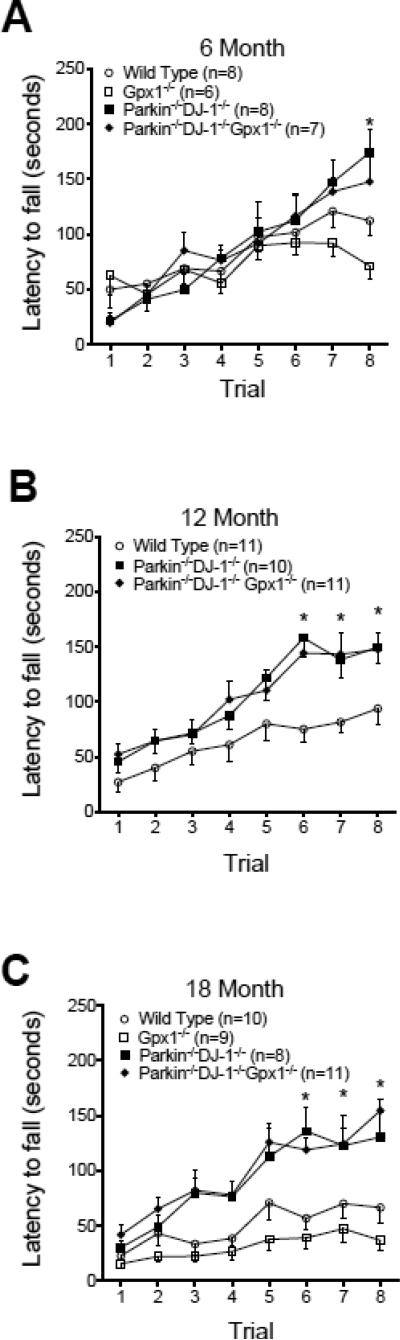
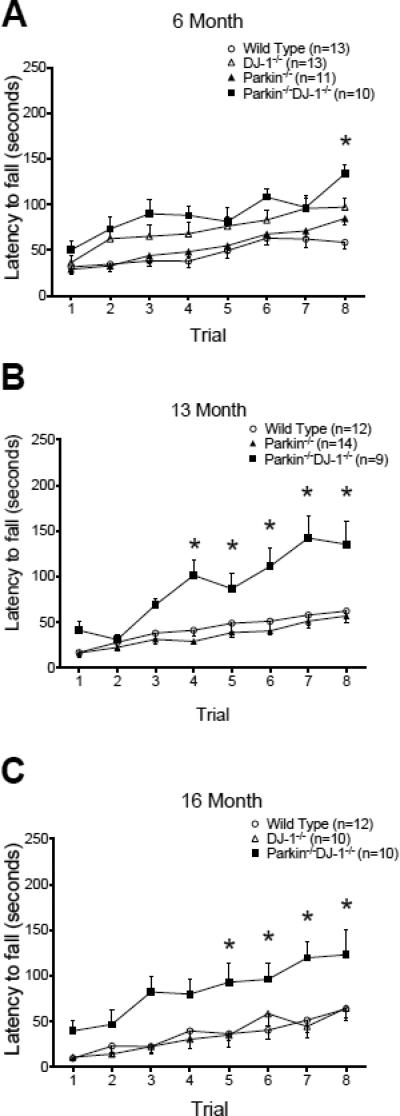
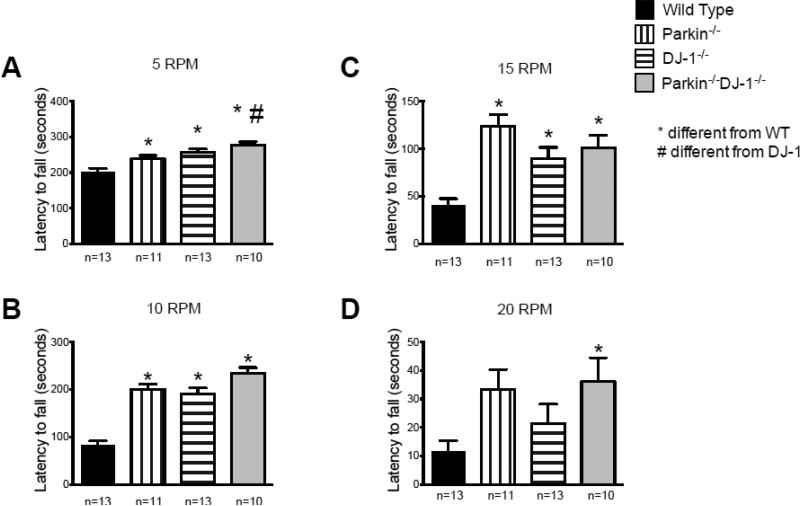
Parkinson's disease (PD) is the second most common neurodegenerative disorder behind Alzheimer's disease. There are currently no therapies proven to halt or slow the progressive neuronal cell loss in PD. A better understanding of the molecular and cellular causes of PD is needed to develop disease-modifying therapies. PD is an age-dependent disease that causes the progressive death of dopamine-producing neurons in the brain. Loss of substantia nigra dopaminergic neurons results in locomotor symptoms such as slowness of movement, tremor, rigidity and postural instability. Abnormalities in other neurotransmitters, such as serotonin, may also be involved in both the motor and non-motor symptoms of PD. Most cases of PD are sporadic but many families show a Mendelian pattern of inherited Parkinsonism and causative mutations have been identified in genes such as Parkin, DJ-1, PINK1, alpha-synuclein and leucine rich repeat kinase 2 (LRRK2). Although the definitive causes of idiopathic PD remain uncertain, the activity of the antioxidant enzyme glutathione peroxidase 1 (Gpx1) is reduced in PD brains and has been shown to be a key determinant of vulnerability to dopaminergic neuron loss in PD animal models. Furthermore, Gpx1 activity decreases with age in human substantia nigra but not rodent substantia nigra. Therefore, we crossed mice deficient for both Parkin and DJ-1 with mice deficient for Gpx1 to test the hypothesis that loss-of-function mutations in Parkin and DJ-1 cause PD by increasing vulnerability to Gpx1 deficiency. Surprisingly, mice lacking Parkin, DJ-1 and Gpx1 have increased striatal dopamine levels in the absence of nigral cell loss compared to wild type, Gpx1(-/-), and Parkin(-/-)DJ-1(-/-) mutant mice. Additionally, Parkin(-/-)DJ-1(-/-) mice exhibit improved rotarod performance and have increased serotonin in the striatum and hippocampus. Stereological analysis indicated that the increased serotonin levels were not due to increased serotonergic projections. The results of our behavioral, neurochemical and immunohistochemical analyses reveal that PD-linked mutations in Parkin and DJ-1 cause dysregulation of neurotransmitter systems beyond the nigrostriatal dopaminergic circuit and that loss-of-function mutations in Parkin and DJ-1 lead to adaptive changes in dopamine and serotonin especially in the context of Gpx1 deficiency.
帕金森病(PD)是仅次于阿尔茨海默病的第二大常见神经退行性疾病。目前尚无被证实可阻止或减缓 PD 中进行性神经元细胞丢失的疗法。为了开发疾病修饰疗法,需要更好地了解 PD 的分子和细胞原因。PD 是一种与年龄相关的疾病,导致大脑中产生多巴胺的神经元进行性死亡。黑质多巴胺能神经元的丧失导致运动症状,如运动缓慢、震颤、僵硬和姿势不稳。其他神经递质(如 5-羟色胺)的异常也可能参与 PD 的运动和非运动症状。大多数 PD 病例为散发性,但许多家族表现出孟德尔遗传帕金森病模式,并已在 Parkin、DJ-1、PINK1、α-突触核蛋白和富含亮氨酸重复激酶 2(LRRK2)等基因中鉴定出致病突变。尽管特发性 PD 的确切原因仍不确定,但 PD 大脑中的抗氧化酶谷胱甘肽过氧化物酶 1(Gpx1)活性降低,并已被证明是 PD 动物模型中多巴胺能神经元丢失易感性的关键决定因素。此外,人类黑质中的 Gpx1 活性随年龄增长而降低,但在啮齿动物黑质中则不会。因此,我们将同时缺乏 Parkin 和 DJ-1 的小鼠与缺乏 Gpx1 的小鼠进行杂交,以测试 Parkin 和 DJ-1 的功能丧失突变是否通过增加对 Gpx1 缺乏的易感性导致 PD 的假设。令人惊讶的是,与野生型、Gpx1(-/-)和 Parkin(-/-)DJ-1(-/-)突变小鼠相比,缺乏 Parkin、DJ-1 和 Gpx1 的小鼠纹状体多巴胺水平升高,而黑质细胞丢失。此外,Parkin(-/-)DJ-1(-/-)小鼠在旋转棒上的表现有所改善,纹状体和海马中的 5-羟色胺增加。体视学分析表明,增加的 5-羟色胺水平不是由于 5-羟色胺能投射增加所致。我们的行为、神经化学和免疫组织化学分析结果表明,Parkin 和 DJ-1 中的 PD 相关突变导致除黑质纹状体多巴胺能回路之外的神经递质系统失调,并且 Parkin 和 DJ-1 的功能丧失突变导致多巴胺和 5-羟色胺的适应性变化,尤其是在 Gpx1 缺乏的情况下。