Laboratoire Evolution, Génomes, Spéciation, CNRS UPR9034/Université Paris-Sud, Gif-sur-Yvette, France.
BMC Genomics. 2013 Oct 11;14:700. doi: 10.1186/1471-2164-14-700.
Insertion Sequences (ISs) and their non-autonomous derivatives (MITEs) are important components of prokaryotic genomes inducing duplication, deletion, rearrangement or lateral gene transfers. Although ISs and MITEs are relatively simple and basic genetic elements, their detection remains a difficult task due to their remarkable sequence diversity. With the advent of high-throughput genome and metagenome sequencing technologies, the development of fast, reliable and sensitive methods of ISs and MITEs detection become an important challenge. So far, almost all studies dealing with prokaryotic transposons have used classical BLAST-based detection methods against reference libraries. Here we introduce alternative methods of detection either taking advantages of the structural properties of the elements (de novo methods) or using an additional library-based method using profile HMM searches.
In this study, we have developed three different work flows dedicated to ISs and MITEs detection: the first two use de novo methods detecting either repeated sequences or presence of Inverted Repeats; the third one use 28 in-house transposase alignment profiles with HMM search methods. We have compared the respective performances of each method using a reference dataset of 30 archaeal and 30 bacterial genomes in addition to simulated and real metagenomes. Compared to a BLAST-based method using ISFinder as library, de novo methods significantly improve ISs and MITEs detection. For example, in the 30 archaeal genomes, we discovered 30 new elements (+20%) in addition to the 141 multi-copies elements already detected by the BLAST approach. Many of the new elements correspond to ISs belonging to unknown or highly divergent families. The total number of MITEs has even doubled with the discovery of elements displaying very limited sequence similarities with their respective autonomous partners (mainly in the Inverted Repeats of the elements). Concerning metagenomes, with the exception of short reads data (<300 bp) for which both techniques seem equally limited, profile HMM searches considerably ameliorate the detection of transposase encoding genes (up to +50%) generating low level of false positives compare to BLAST-based methods.
Compared to classical BLAST-based methods, the sensitivity of de novo and profile HMM methods developed in this study allow a better and more reliable detection of transposons in prokaryotic genomes and metagenomes. We believed that future studies implying ISs and MITEs identification in genomic data should combine at least one de novo and one library-based method, with optimal results obtained by running the two de novo methods in addition to a library-based search. For metagenomic data, profile HMM search should be favored, a BLAST-based step is only useful to the final annotation into groups and families.
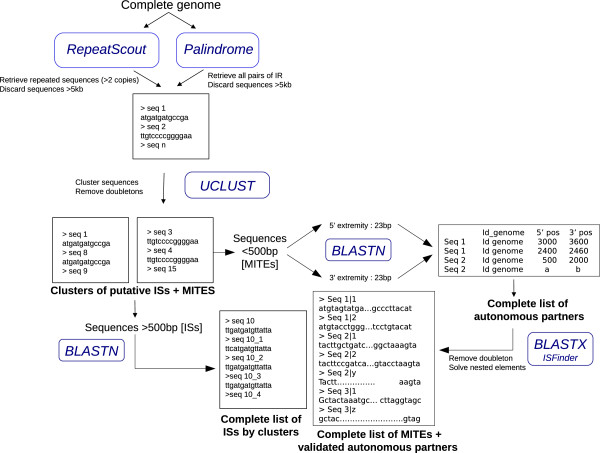
插入序列(ISs)及其非自主衍生物(MITEs)是诱导细菌基因组中重复、缺失、重排或水平基因转移的重要组成部分。尽管 ISs 和 MITEs 是相对简单和基本的遗传元件,但由于其显著的序列多样性,它们的检测仍然是一项具有挑战性的任务。随着高通量基因组和宏基因组测序技术的出现,开发快速、可靠和敏感的 ISs 和 MITEs 检测方法成为一个重要的挑战。到目前为止,几乎所有涉及原核转座子的研究都使用了针对参考文库的基于经典 BLAST 的检测方法。在这里,我们介绍了利用元件结构特性(从头开始方法)或使用基于附加文库的方法进行检测的替代方法,该方法使用轮廓 HMM 搜索。
在这项研究中,我们开发了三种专门用于 ISs 和 MITEs 检测的不同工作流程:前两种方法使用从头开始的方法检测重复序列或倒置重复的存在;第三种方法使用 28 个内部转座酶比对轮廓进行 HMM 搜索方法。我们使用 30 个古细菌和 30 个细菌基因组的参考数据集以及模拟和真实宏基因组对每种方法的性能进行了比较。与基于 ISFinder 的 BLAST 方法相比,从头开始的方法显著提高了 ISs 和 MITEs 的检测能力。例如,在 30 个古细菌基因组中,除了 BLAST 方法已经检测到的 141 个多拷贝元件外,我们还发现了 30 个新元件(+20%)。许多新元件属于未知或高度分化的家族的 ISs。通过发现与自主伙伴具有非常有限序列相似性的元件(主要在元件的倒置重复中),MITE 的总数甚至翻了一番。关于宏基因组,除了两种技术似乎同样受到限制的短读长数据(<300 bp)外,轮廓 HMM 搜索极大地改善了转座酶编码基因的检测(高达+50%),与基于 BLAST 的方法相比产生的假阳性水平较低。
与经典的基于 BLAST 的方法相比,本研究中开发的从头开始和轮廓 HMM 方法的敏感性允许在原核基因组和宏基因组中更好、更可靠地检测转座子。我们认为,未来涉及基因组数据中转座子识别的研究应至少结合一种从头开始的方法和一种基于文库的方法,通过运行两种从头开始的方法并结合基于文库的搜索,可以获得最佳结果。对于宏基因组数据,应首选轮廓 HMM 搜索,基于 BLAST 的步骤仅对最终的分组和家族注释有用。

