Department of Molecular Medicine, University of Padova, Padova, Italy.
BMC Infect Dis. 2013 Nov 19;13:554. doi: 10.1186/1471-2334-13-554.
Next generation sequencing (NGS) is being increasingly used for the detection and characterization of pathogens during outbreaks. This technology allows rapid sequencing of pathogen full genomes, useful not only for accurate genotyping and molecular epidemiology, but also for identification of drug resistance and virulence traits.
In this study, an approach based on whole genome sequencing by NGS, comparative genomics, and gene function prediction was set up and retrospectively applied for the investigation of two N. meningitidis serogroup C isolates collected from a cluster of meningococcal disease, characterized by a high fatality rate.
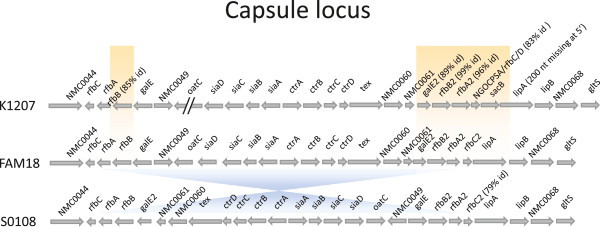
According to conventional molecular typing methods, all the isolates had the same typing results and were classified as outbreak isolates within the same N. meningitidis sequence type ST-11, while full genome sequencing demonstrated subtle genetic differences between the isolates. Looking for these specific regions by means of 9 PCR and cycle sequencing assays in other 7 isolates allowed distinguishing outbreak cases from unrelated cases. Comparative genomics and gene function prediction analyses between outbreak isolates and a set of reference N. meningitidis genomes led to the identification of differences in gene content that could be relevant for pathogenesis. Most genetic changes occurred in the capsule locus and were consistent with recombination and horizontal acquisition of a set of genes involved in capsule biosynthesis.
This study showed the added value given by whole genome sequencing by NGS over conventional sequence-based typing methods in the investigation of an outbreak. Routine application of this technology in clinical microbiology will significantly improve methods for molecular epidemiology and surveillance of infectious disease and provide a bulk of data useful to improve our understanding of pathogens biology.
下一代测序(NGS)技术越来越多地用于暴发期间病原体的检测和特征分析。这项技术不仅可用于病原体全基因组的快速测序,以实现准确的基因分型和分子流行病学,还可用于鉴定耐药性和毒力特征。
本研究建立了一种基于 NGS 全基因组测序、比较基因组学和基因功能预测的方法,并回顾性地应用于调查两起脑膜炎奈瑟菌 C 群分离株,这些分离株来自一组脑膜炎球菌病,其病死率高。
根据传统的分子分型方法,所有分离株的结果均相同,且均归类为同一脑膜炎奈瑟菌序列型 ST-11 内的暴发分离株,而全基因组测序显示分离株之间存在细微的遗传差异。通过 9 种 PCR 和循环测序试验在其他 7 株分离株中寻找这些特定区域,可将暴发病例与无关病例区分开来。对暴发分离株与一组参考脑膜炎奈瑟菌基因组进行比较基因组学和基因功能预测分析,确定了与发病机制相关的基因内容差异。大多数遗传变化发生在荚膜基因座,与荚膜生物合成相关基因的重组和水平获得一致。
本研究表明,NGS 全基因组测序在暴发调查中比传统的基于序列的分型方法具有更大的附加价值。在临床微生物学中常规应用这项技术将极大地改善传染病的分子流行病学和监测方法,并提供大量有助于提高我们对病原体生物学理解的数据。