Vellore Nadeem A, Baron Riccardo
Department of Medicinal Chemistry, College of Pharmacy, and The Henry Eyring Center for Theoretical Chemistry, The University of Utah, Salt Lake City, UT 84112-5820, USA.
BMC Biophys. 2013 Nov 25;6(1):15. doi: 10.1186/2046-1682-6-15.
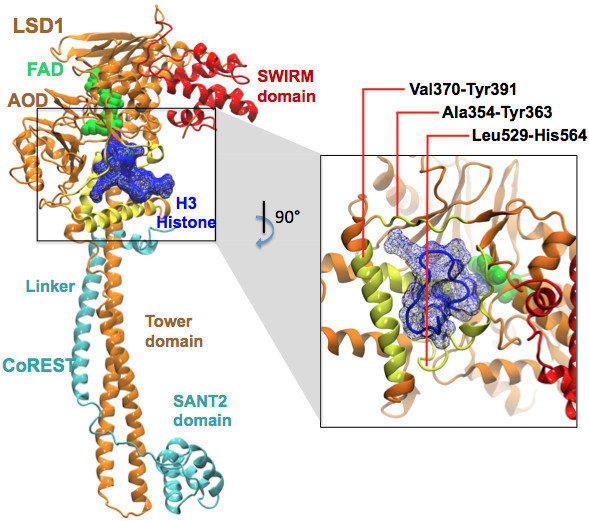
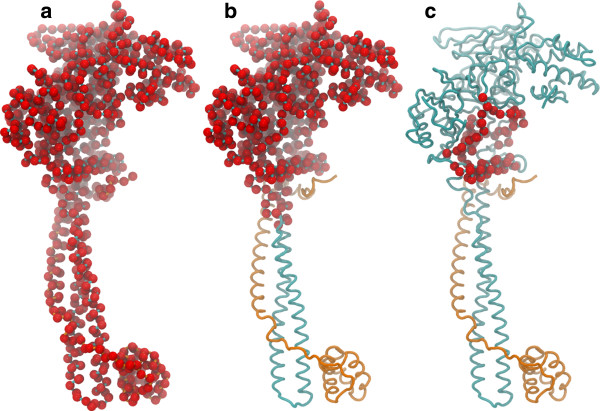
Lysine Specific Demethylase (LSD1 or KDM1A) in complex with its co-repressor protein CoREST catalyzes the demethylation of the H3 histone N-terminal tail and is currently one of the most promising epigenetic targets for drug discovery against cancer and neurodegenerative diseases. Models of non-covalent binding, such as lock and key, induced-fit, and conformational selection could help explaining the molecular mechanism of LSD1/CoREST-H3-histone association, thus guiding drug discovery and design efforts. Here, we quantify the extent to which LSD1/CoREST substrate binding is consistent with these hypothetical models using LSD1/CoREST conformational ensembles obtained through extensive explicit solvent molecular dynamics (MD) simulations.
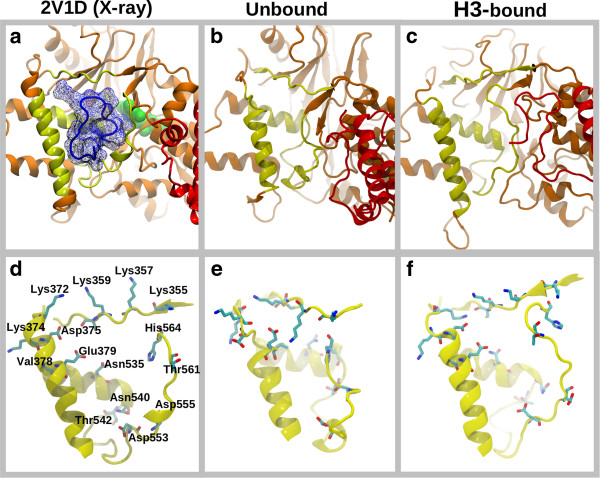
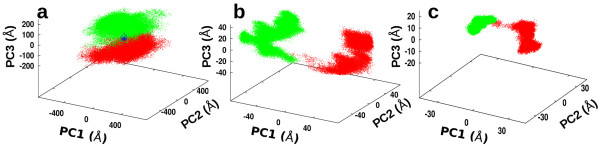
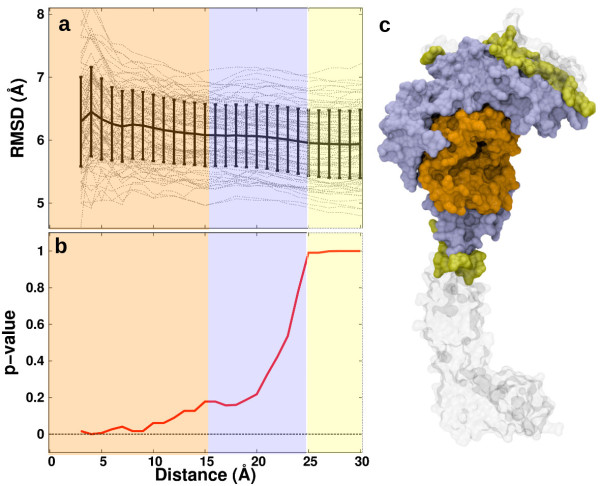
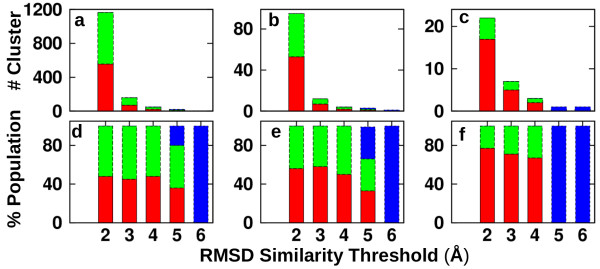
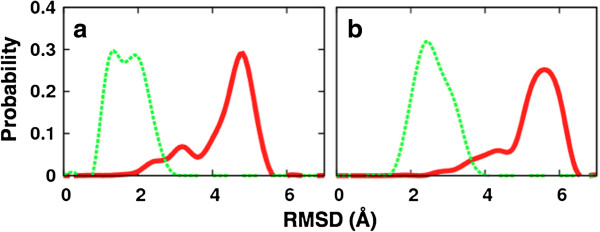
We find that an induced-fit model is the most representative of LSD1/CoREST-H3-histone non-covalent binding and accounts for the local conformational changes occurring in the H3-histone binding site. We also show that conformational selection - despite in principle not ruled out by this finding - is minimal, and only relevant when global properties are considered, e.g. the nanoscale motion of the LSD1/CoREST clamp.
The induced-fit mechanism revealed by our MD simulation study will aid the inclusion of protein dynamics for the discovery and design of LSD1 inhibitors targeting the H3-histone binding region. On a general basis, our study indicates the importance of using multiple metrics or selection schemes when testing alternative hypothetical mechanistic models of non-covalent binding.
赖氨酸特异性去甲基化酶(LSD1或KDM1A)与其共抑制蛋白CoREST形成复合物,催化H3组蛋白N端尾巴的去甲基化,目前是抗癌和神经退行性疾病药物研发中最有前景的表观遗传靶点之一。非共价结合模型,如锁钥模型、诱导契合模型和构象选择模型,有助于解释LSD1/CoREST与H3组蛋白结合的分子机制,从而指导药物研发工作。在此,我们使用通过广泛的显式溶剂分子动力学(MD)模拟获得的LSD1/CoREST构象集合,量化LSD1/CoREST底物结合与这些假设模型的符合程度。
我们发现诱导契合模型最能代表LSD1/CoREST与H3组蛋白的非共价结合,并解释了H3组蛋白结合位点发生的局部构象变化。我们还表明,构象选择——尽管原则上不被这一发现排除——作用极小,仅在考虑全局性质时相关,例如LSD1/CoREST夹子的纳米级运动。
我们的MD模拟研究揭示的诱导契合机制将有助于在发现和设计靶向H3组蛋白结合区域的LSD1抑制剂时纳入蛋白质动力学。一般而言,我们的研究表明在测试非共价结合的替代假设机制模型时使用多种指标或选择方案的重要性。