Department of Biostatistics and Computational Biology, Dana-Farber Cancer Institute and Harvard School of Public Health, Boston, Massachusetts 02115, USA.
Department of Medical Oncology, Dana-Farber Cancer Institute and Harvard Medical School, Boston, Massachusetts 02115, USA.
Nat Methods. 2014 Jan;11(1):73-78. doi: 10.1038/nmeth.2762. Epub 2013 Dec 8.
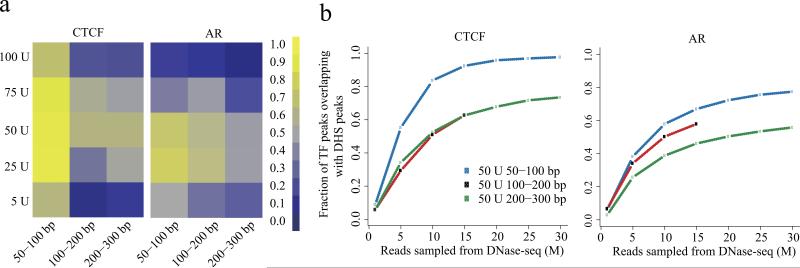
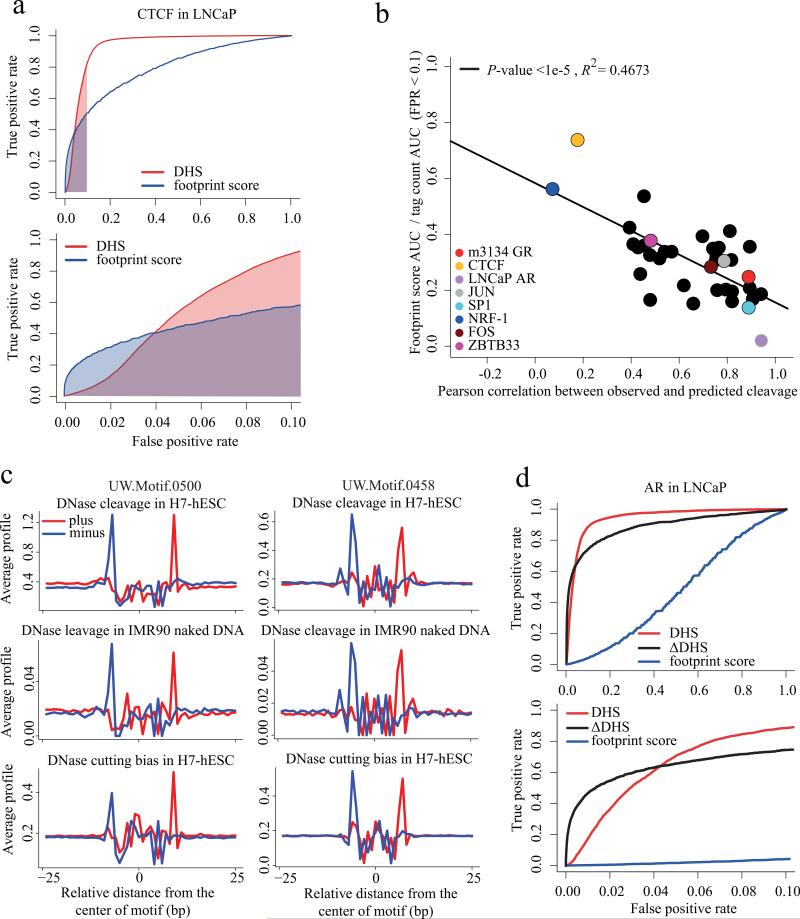
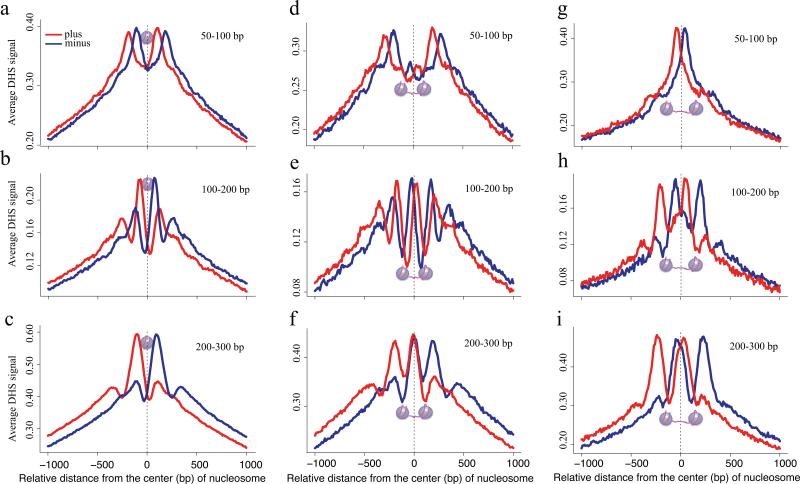
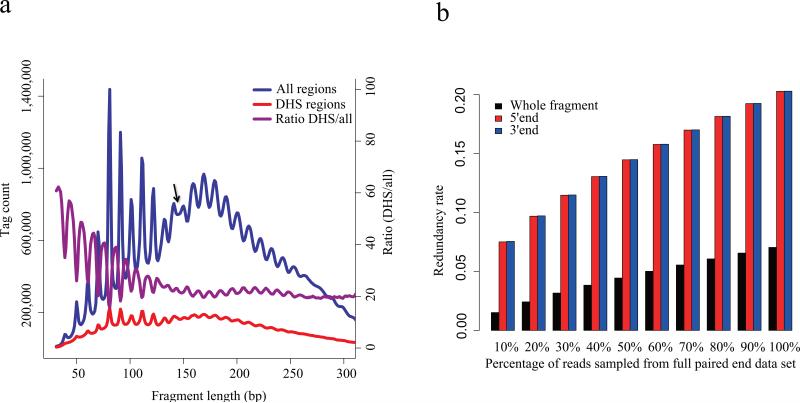
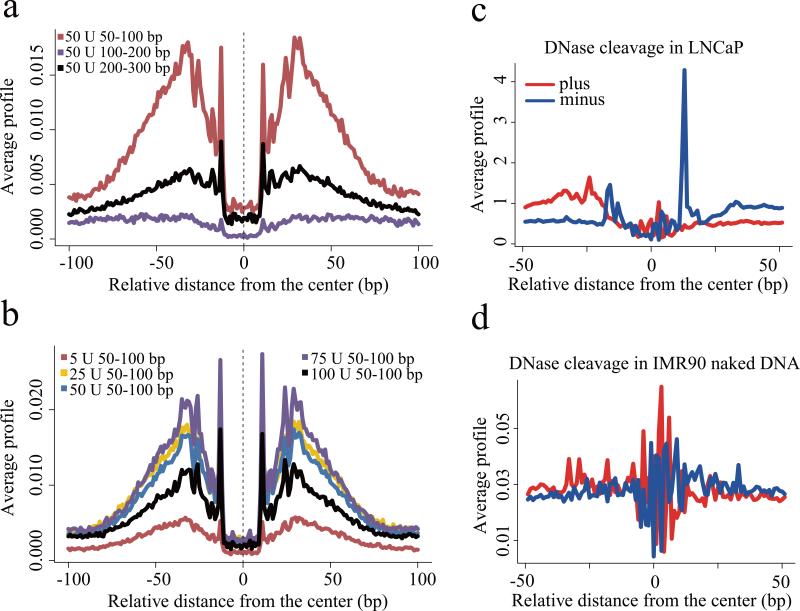
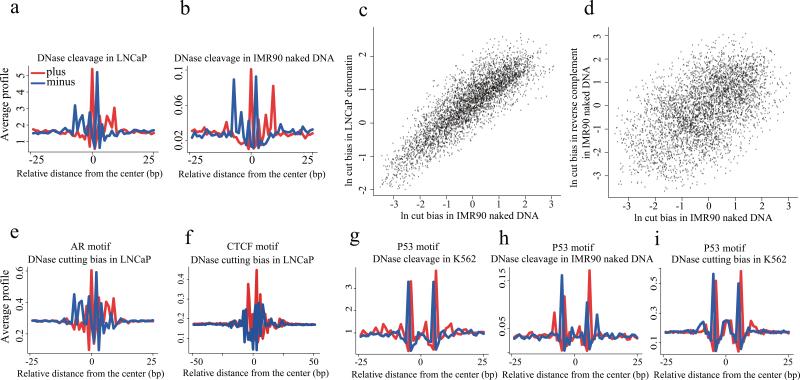
Sequencing of DNase I hypersensitive sites (DNase-seq) is a powerful technique for identifying cis-regulatory elements across the genome. We studied the key experimental parameters to optimize performance of DNase-seq. Sequencing short fragments of 50-100 base pairs (bp) that accumulate in long internucleosome linker regions was more efficient for identifying transcription factor binding sites compared to sequencing longer fragments. We also assessed the potential of DNase-seq to predict transcription factor occupancy via generation of nucleotide-resolution transcription factor footprints. In modeling the sequence-specific DNase I cutting bias, we found a strong effect that varied over more than two orders of magnitude. This indicates that the nucleotide-resolution cleavage patterns at many transcription factor binding sites are derived from intrinsic DNase I cleavage bias rather than from specific protein-DNA interactions. In contrast, quantitative comparison of DNase I hypersensitivity between states can predict transcription factor occupancy associated with particular biological perturbations.
DNase I 超敏位点测序(DNase-seq)是一种强大的技术,可用于鉴定整个基因组中的顺式调控元件。我们研究了关键的实验参数,以优化 DNase-seq 的性能。与测序较长片段相比,对 50-100 个碱基对(bp)的短片段进行测序,更有利于鉴定转录因子结合位点,因为这些短片段在长核小体连接区积累。我们还通过生成核苷酸分辨率的转录因子足迹,评估了 DNase-seq 预测转录因子占据的潜力。在对序列特异性 DNase I 切割偏倚进行建模时,我们发现了一个强烈的影响,其变化超过两个数量级。这表明,许多转录因子结合位点的核苷酸分辨率切割模式源自内在的 DNase I 切割偏倚,而不是来自特定的蛋白质-DNA 相互作用。相比之下,对不同状态下的 DNase I 超敏性进行定量比较,可以预测与特定生物学扰动相关的转录因子占据。