Hu Yan, Horlbeck Max A, Zhang Ruochi, Ma Sai, Shrestha Rojesh, Kartha Vinay K, Duarte Fabiana M, Hock Conrad, Savage Rachel E, Labade Ajay, Kletzien Heidi, Meliki Alia, Castillo Andrew, Durand Neva C, Mattei Eugenio, Anderson Lauren J, Tay Tristan, Earl Andrew S, Shoresh Noam, Epstein Charles B, Wagers Amy J, Buenrostro Jason D
Gene Regulation Observatory, Broad Institute of MIT and Harvard, Cambridge, MA, USA.
Department of Stem Cell and Regenerative Biology, Harvard University, Cambridge, MA, USA.
Nature. 2025 Feb;638(8051):779-786. doi: 10.1038/s41586-024-08443-4. Epub 2025 Jan 22.
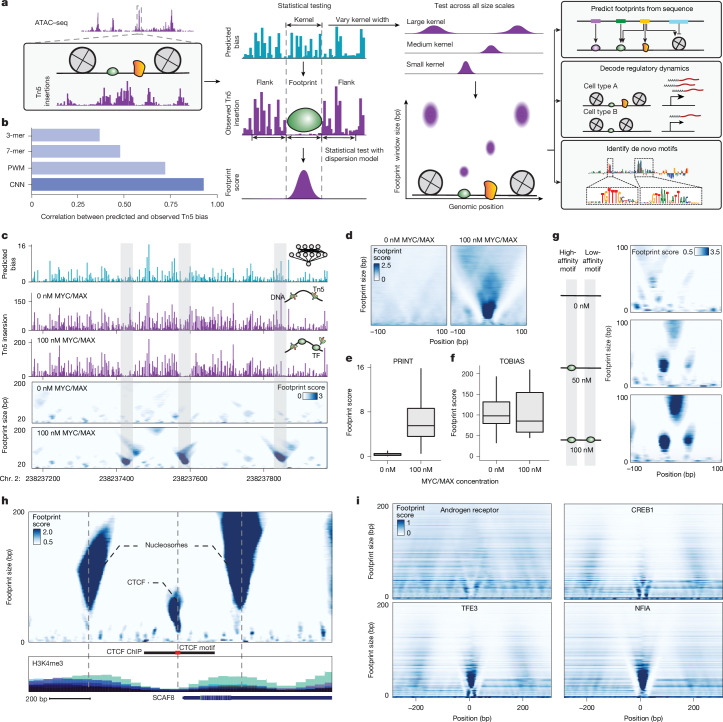
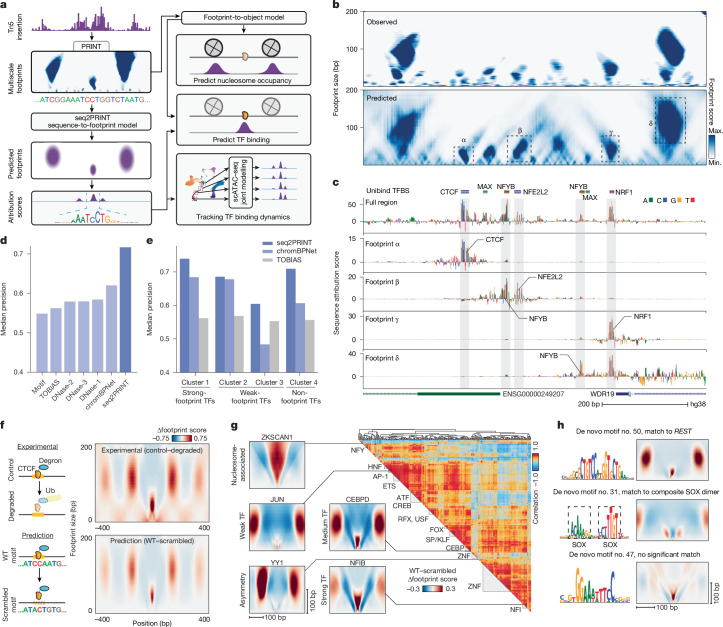
Cis-regulatory elements (CREs) control gene expression and are dynamic in their structure and function, reflecting changes in the composition of diverse effector proteins over time. However, methods for measuring the organization of effector proteins at CREs across the genome are limited, hampering efforts to connect CRE structure to their function in cell fate and disease. Here we developed PRINT, a computational method that identifies footprints of DNA-protein interactions from bulk and single-cell chromatin accessibility data across multiple scales of protein size. Using these multiscale footprints, we created the seq2PRINT framework, which uses deep learning to allow precise inference of transcription factor and nucleosome binding and interprets regulatory logic at CREs. Applying seq2PRINT to single-cell chromatin accessibility data from human bone marrow, we observe sequential establishment and widening of CREs centred on pioneer factors across haematopoiesis. We further discover age-associated alterations in the structure of CREs in murine haematopoietic stem cells, including widespread reduction of nucleosome footprints and gain of de novo identified Ets composite motifs. Collectively, we establish a method for obtaining rich insights into DNA-binding protein dynamics from chromatin accessibility data, and reveal the architecture of regulatory elements across differentiation and ageing.
顺式调控元件(CREs)控制基因表达,其结构和功能具有动态性,反映了不同效应蛋白组成随时间的变化。然而,测量全基因组中CREs处效应蛋白组织方式的方法有限,这阻碍了将CRE结构与其在细胞命运和疾病中的功能联系起来的研究工作。在此,我们开发了PRINT,这是一种计算方法,可从跨多种蛋白质大小尺度的批量和单细胞染色质可及性数据中识别DNA-蛋白质相互作用的足迹。利用这些多尺度足迹,我们创建了seq2PRINT框架,该框架使用深度学习来精确推断转录因子和核小体的结合,并解读CREs处的调控逻辑。将seq2PRINT应用于来自人类骨髓的单细胞染色质可及性数据,我们观察到在造血过程中以先驱因子为中心的CREs的顺序建立和扩展。我们进一步发现小鼠造血干细胞中CREs结构与年龄相关的改变,包括核小体足迹的广泛减少和新鉴定的Ets复合基序的增加。总体而言,我们建立了一种从染色质可及性数据中深入了解DNA结合蛋白动态的方法,并揭示了跨分化和衰老过程中调控元件的结构。