Department of Bioengineering, University of Illinois at Urbana-Champaign, Urbana, Illinois, United States of America.
PLoS Comput Biol. 2013;9(12):e1003367. doi: 10.1371/journal.pcbi.1003367. Epub 2013 Dec 5.
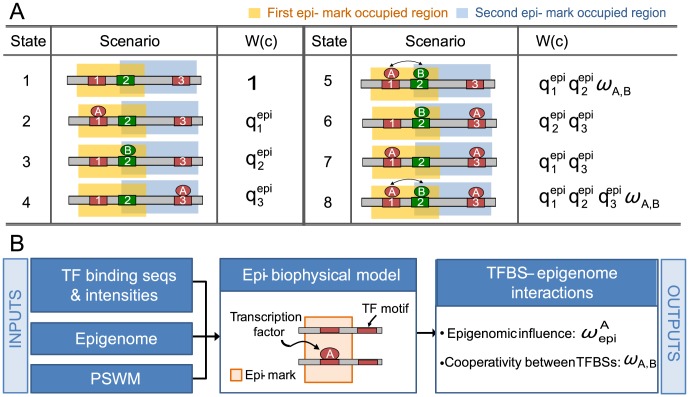
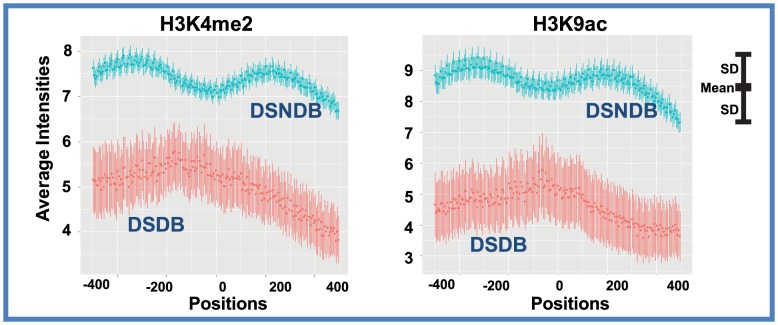
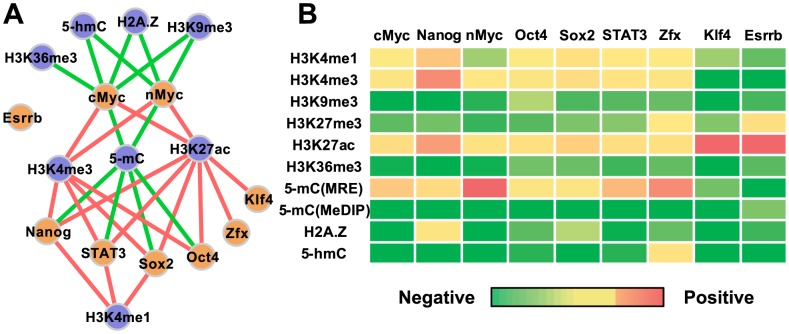
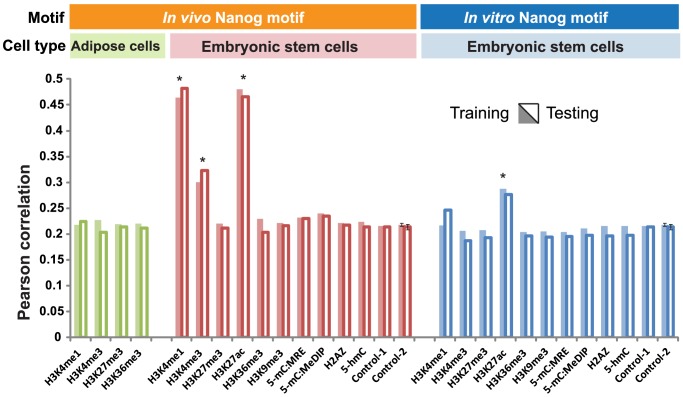
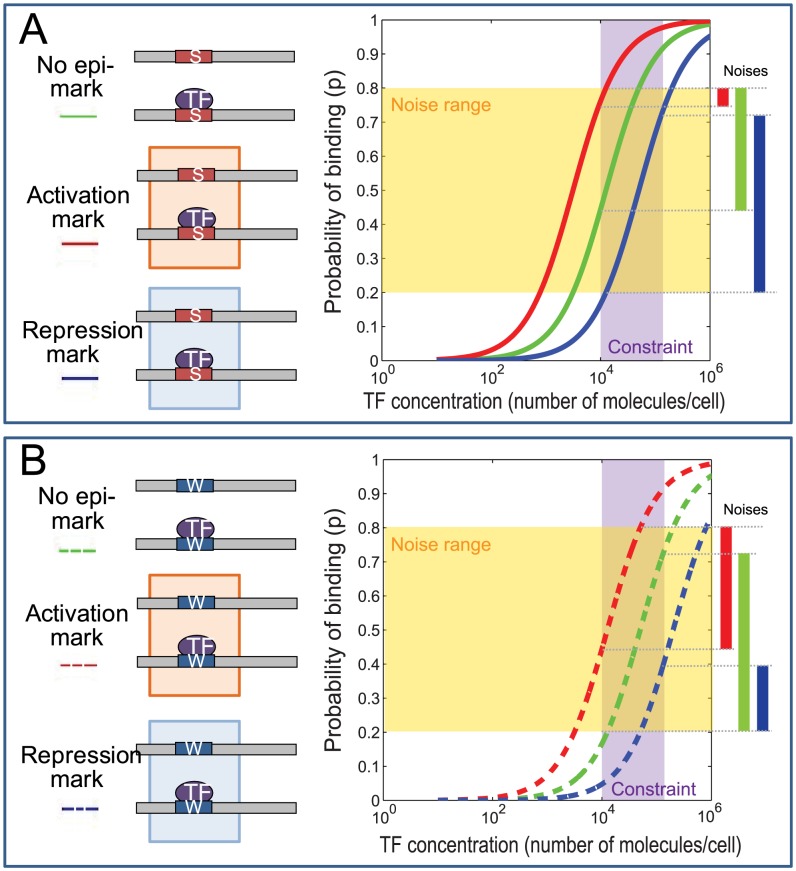
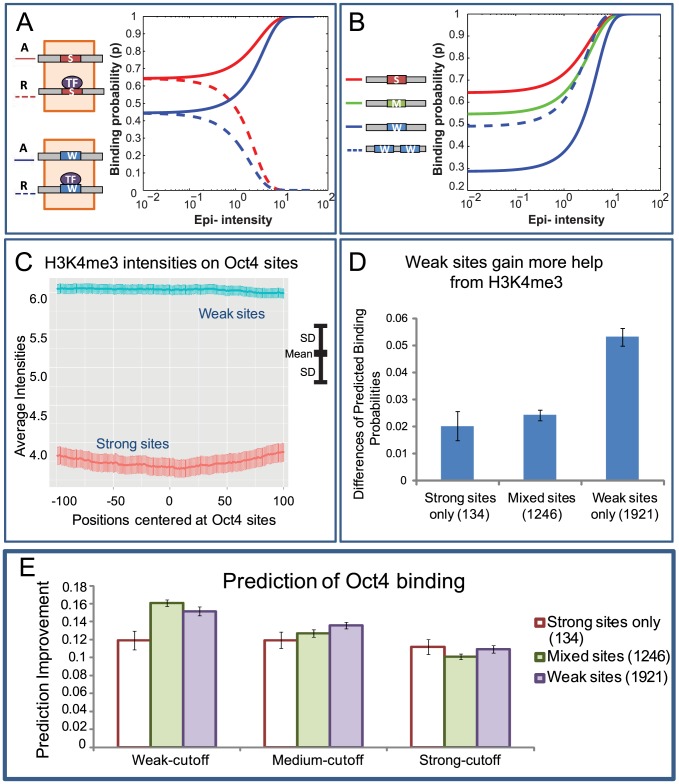
Despite explosive growth in genomic datasets, the methods for studying epigenomic mechanisms of gene regulation remain primitive. Here we present a model-based approach to systematically analyze the epigenomic functions in modulating transcription factor-DNA binding. Based on the first principles of statistical mechanics, this model considers the interactions between epigenomic modifications and a cis-regulatory module, which contains multiple binding sites arranged in any configurations. We compiled a comprehensive epigenomic dataset in mouse embryonic stem (mES) cells, including DNA methylation (MeDIP-seq and MRE-seq), DNA hydroxymethylation (5-hmC-seq), and histone modifications (ChIP-seq). We discovered correlations of transcription factors (TFs) for specific combinations of epigenomic modifications, which we term epigenomic motifs. Epigenomic motifs explained why some TFs appeared to have different DNA binding motifs derived from in vivo (ChIP-seq) and in vitro experiments. Theoretical analyses suggested that the epigenome can modulate transcriptional noise and boost the cooperativity of weak TF binding sites. ChIP-seq data suggested that epigenomic boost of binding affinities in weak TF binding sites can function in mES cells. We showed in theory that the epigenome should suppress the TF binding differences on SNP-containing binding sites in two people. Using personal data, we identified strong associations between H3K4me2/H3K9ac and the degree of personal differences in NFκB binding in SNP-containing binding sites, which may explain why some SNPs introduce much smaller personal variations on TF binding than other SNPs. In summary, this model presents a powerful approach to analyze the functions of epigenomic modifications. This model was implemented into an open source program APEG (Affinity Prediction by Epigenome and Genome, http://systemsbio.ucsd.edu/apeg).
尽管基因组数据集呈爆炸式增长,但研究基因调控的表观遗传机制的方法仍然很原始。在这里,我们提出了一种基于模型的方法来系统地分析调节转录因子-DNA 结合的表观遗传功能。基于统计力学的基本原理,该模型考虑了表观遗传修饰与顺式调控模块之间的相互作用,其中包含以任意构型排列的多个结合位点。我们在小鼠胚胎干细胞 (mES) 细胞中编译了一个全面的表观基因组数据集,包括 DNA 甲基化 (MeDIP-seq 和 MRE-seq)、DNA 羟甲基化 (5-hmC-seq) 和组蛋白修饰 (ChIP-seq)。我们发现了特定组合的表观遗传修饰与转录因子 (TFs) 之间的相关性,我们将其称为表观遗传基序。表观遗传基序解释了为什么一些 TF 似乎具有不同的 DNA 结合基序,这些基序源自体内 (ChIP-seq) 和体外实验。理论分析表明,表观基因组可以调节转录噪声并增强弱 TF 结合位点的协同作用。ChIP-seq 数据表明,在 mES 细胞中,弱 TF 结合位点的表观遗传增强结合亲和力可以发挥作用。我们从理论上表明,在含有 SNP 的结合位点上,表观基因组应该抑制两个人之间 TF 结合差异。使用个人数据,我们确定了 H3K4me2/H3K9ac 与 NFκB 结合在 SNP 结合位点上的个人差异程度之间的强烈关联,这可能解释了为什么一些 SNP 对 TF 结合的个人差异影响比其他 SNP 小得多。总之,该模型为分析表观遗传修饰的功能提供了一种强大的方法。该模型已被实现为一个开源程序 APEG(通过表观基因组和基因组预测亲和力,http://systemsbio.ucsd.edu/apeg)。