Rattray Ivan, Smith Edward J, Crum William R, Walker Thomas A, Gale Richard, Bates Gillian P, Modo Michel
King's College London, Institute of Psychiatry, Department of Neuroscience, London, United Kingdom.
King's College London School of Medicine, Department of Medical and Molecular Genetics, Guy's Hospital, London, United Kingdom.
PLoS One. 2017 Jan 18;12(1):e0168556. doi: 10.1371/journal.pone.0168556. eCollection 2017.
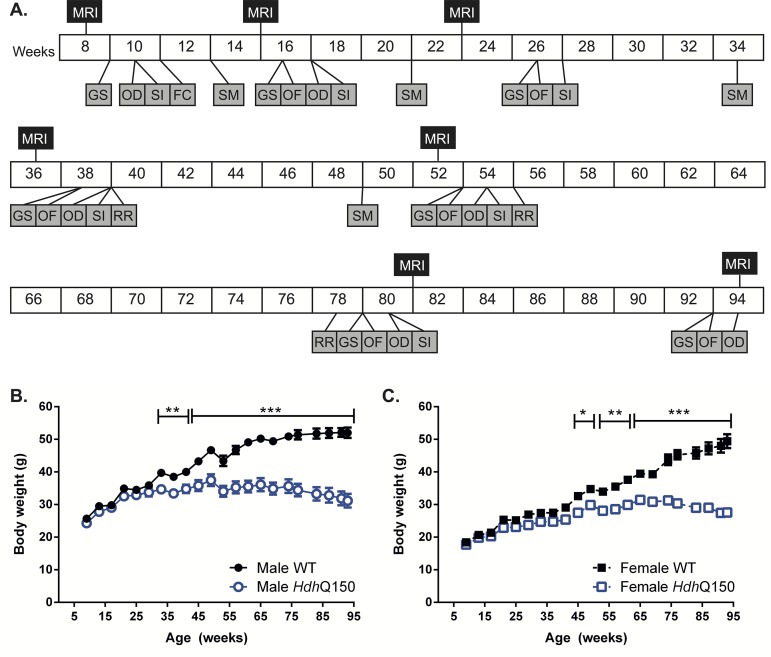
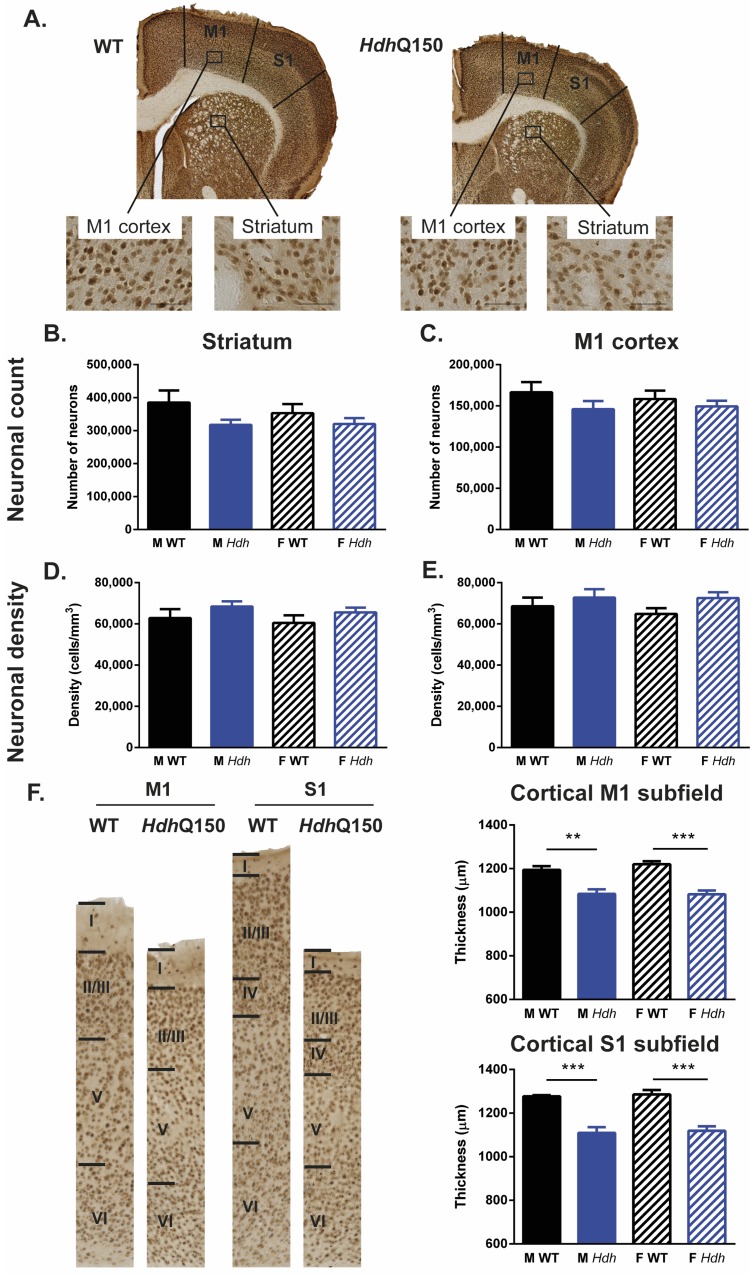
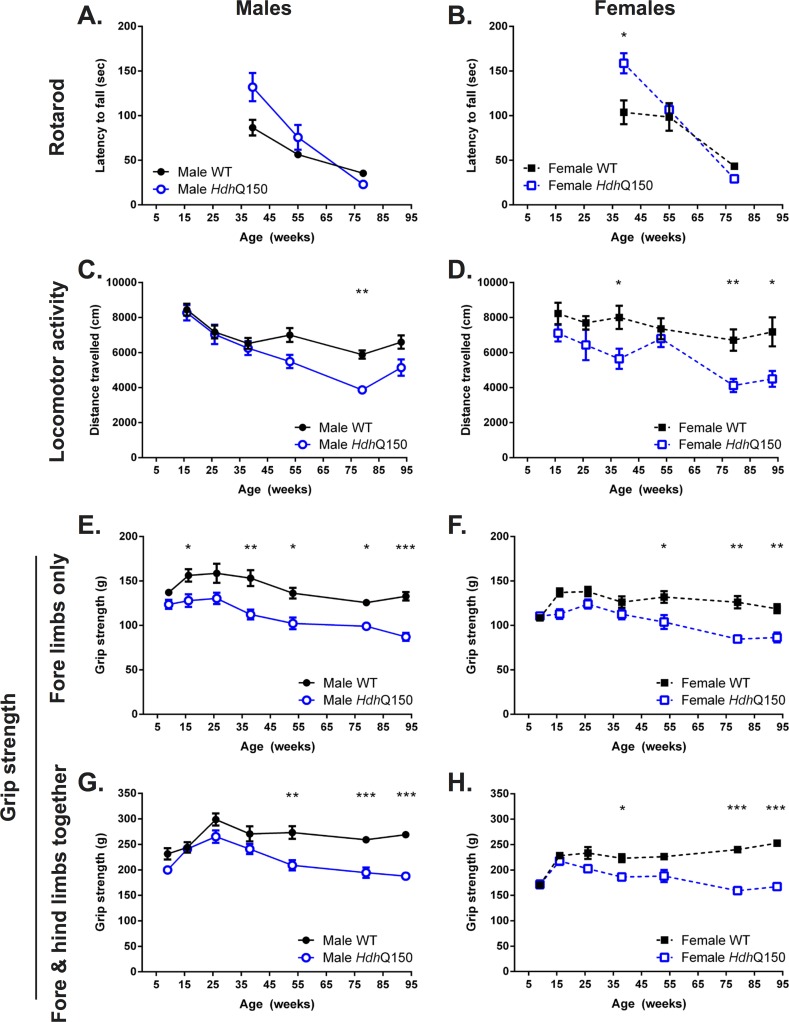
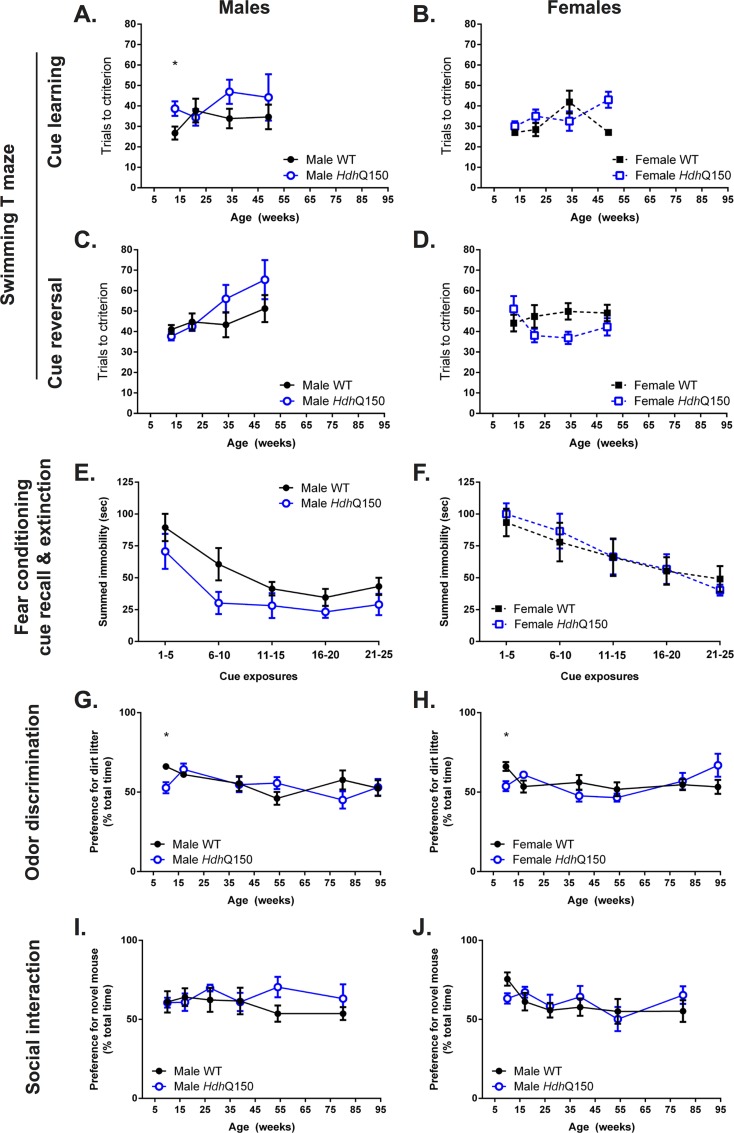
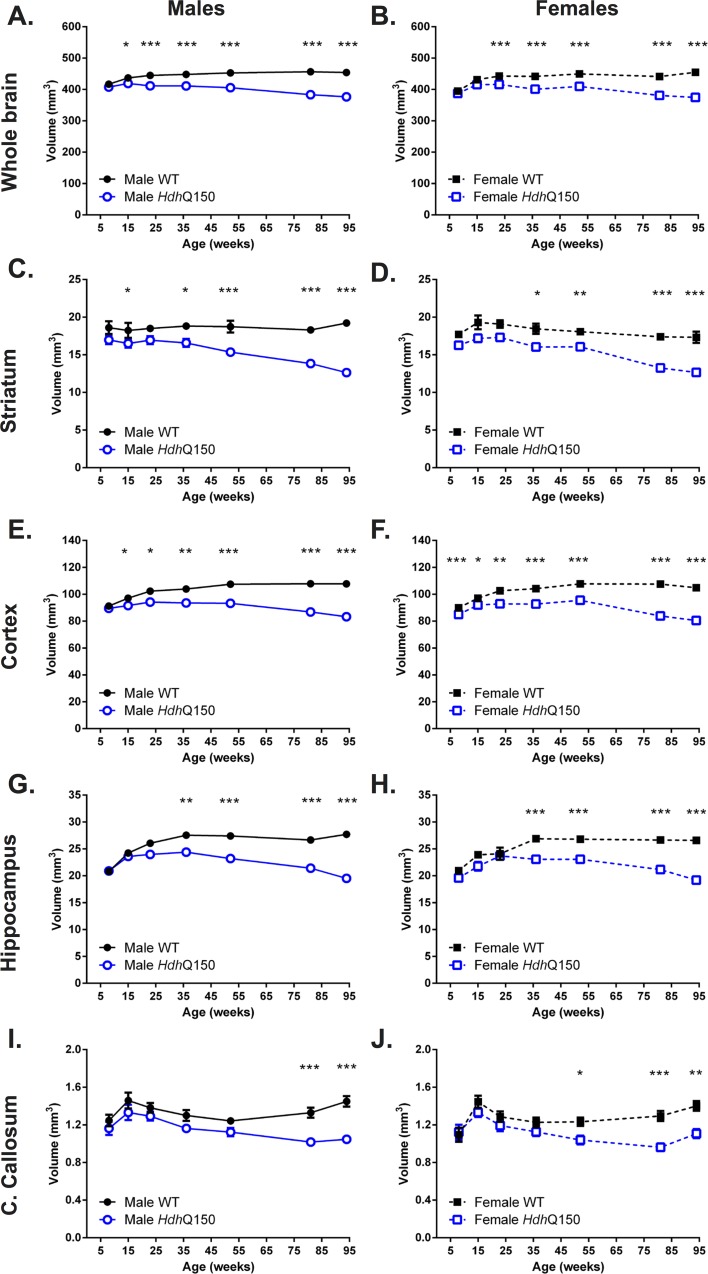
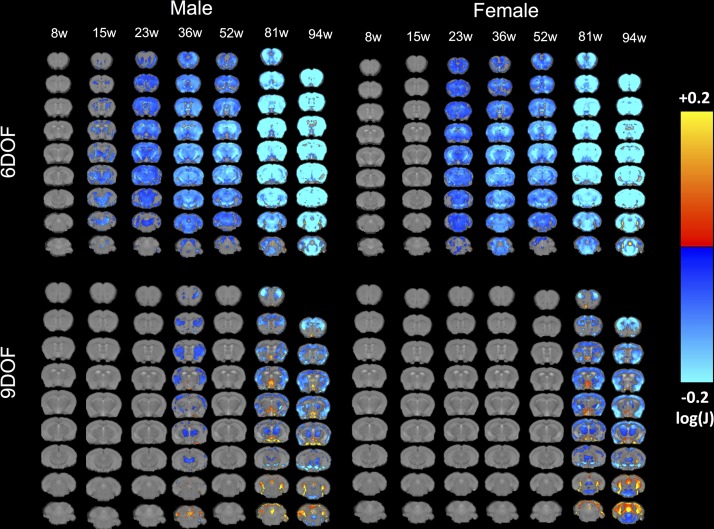
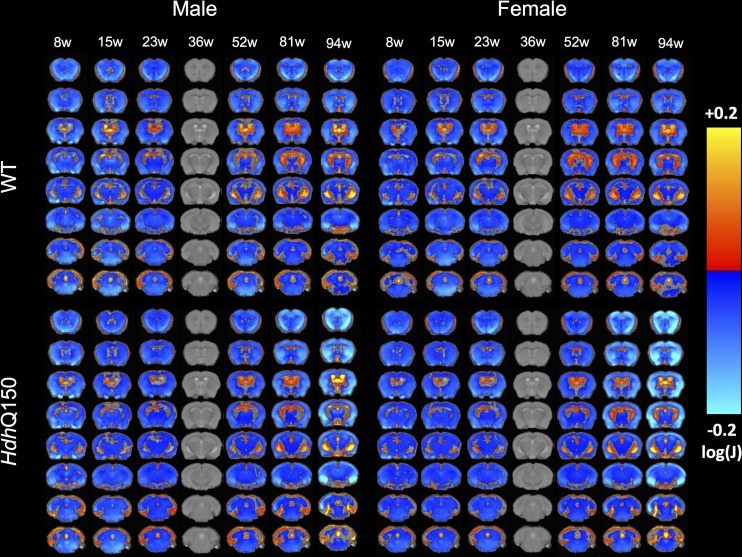
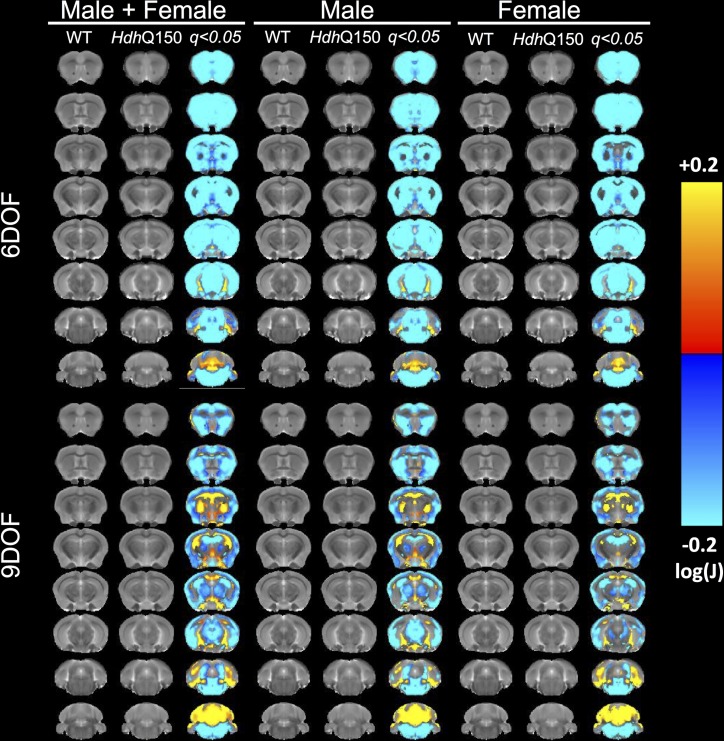
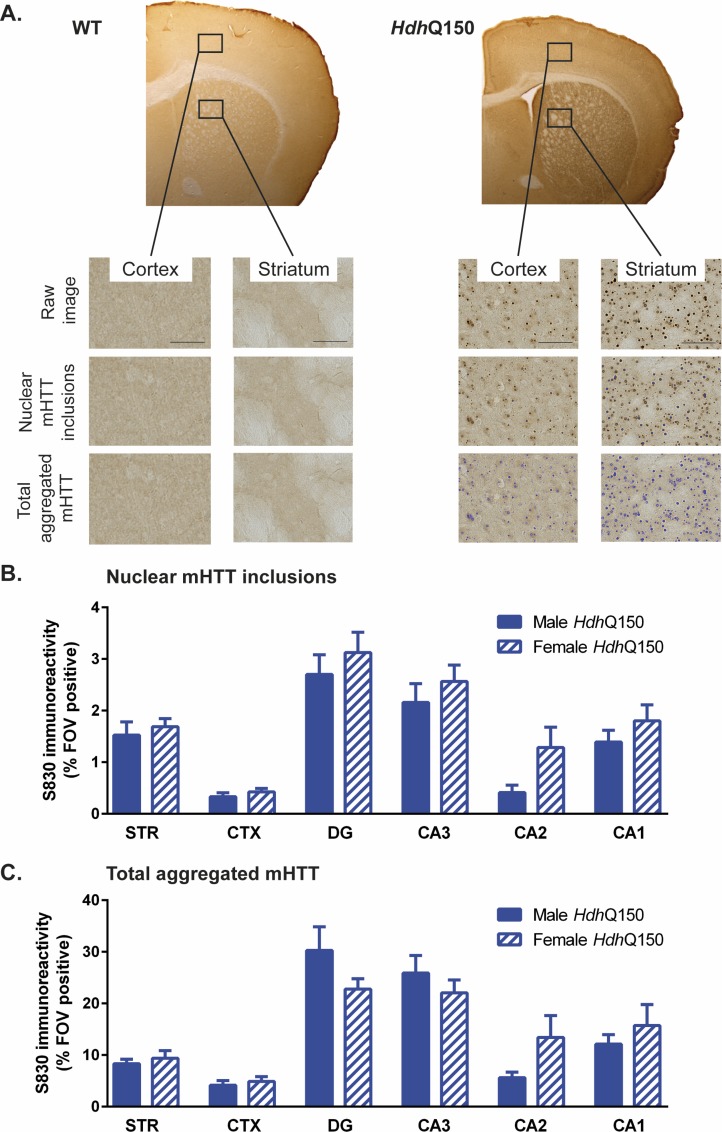
A variety of mouse models have been developed that express mutant huntingtin (mHTT) leading to aggregates and inclusions that model the molecular pathology observed in Huntington's disease. Here we show that although homozygous HdhQ150 knock-in mice developed motor impairments (rotarod, locomotor activity, grip strength) by 36 weeks of age, cognitive dysfunction (swimming T maze, fear conditioning, odor discrimination, social interaction) was not evident by 94 weeks. Concomitant to behavioral assessments, T2-weighted MRI volume measurements indicated a slower striatal growth with a significant difference between wild type (WT) and HdhQ150 mice being present even at 15 weeks. Indeed, MRI indicated significant volumetric changes prior to the emergence of the "clinical horizon" of motor impairments at 36 weeks of age. A striatal decrease of 27% was observed over 94 weeks with cortex (12%) and hippocampus (21%) also indicating significant atrophy. A hypothesis-free analysis using tensor-based morphometry highlighted further regions undergoing atrophy by contrasting brain growth and regional neurodegeneration. Histology revealed the widespread presence of mHTT aggregates and cellular inclusions. However, there was little evidence of correlations between these outcome measures, potentially indicating that other factors are important in the causal cascade linking the molecular pathology to the emergence of behavioral impairments. In conclusion, the HdhQ150 mouse model replicates many aspects of the human condition, including an extended pre-manifest period prior to the emergence of motor impairments.
已经开发出多种表达突变型亨廷顿蛋白(mHTT)的小鼠模型,这些模型会形成聚集体和包涵体,模拟在亨廷顿舞蹈病中观察到的分子病理学特征。在此我们表明,虽然纯合HdhQ150基因敲入小鼠在36周龄时出现了运动障碍(转棒试验、运动活动、握力),但在94周龄时认知功能障碍(游泳T迷宫试验、恐惧条件反射、气味辨别、社交互动)并不明显。与行为评估同时进行的是,T2加权MRI体积测量表明纹状体生长较慢,甚至在15周龄时野生型(WT)和HdhQ150小鼠之间就存在显著差异。事实上,MRI显示在36周龄运动障碍的“临床症状出现之前”就有显著的体积变化。在94周的时间里,纹状体体积减少了27%,皮质(12%)和海马体(21%)也显示出显著萎缩。使用基于张量的形态测量法进行的无假设分析通过对比脑生长和区域神经退行性变,进一步突出了发生萎缩的区域。组织学检查发现mHTT聚集体和细胞包涵体广泛存在。然而,几乎没有证据表明这些结果指标之间存在相关性,这可能表明在将分子病理学与行为障碍出现联系起来的因果级联反应中,其他因素也很重要。总之,HdhQ150小鼠模型复制了人类疾病的许多方面,包括在运动障碍出现之前有一个延长的临床前期。