Science for Life Laboratory, School of Biotechnology, Division of Gene Technology, Royal Institute of Technology, Stockholm, Sweden.
Science for Life Laboratory, School of Biotechnology, Division of Gene Technology, Royal Institute of Technology, Stockholm, Sweden ; Department of Forensic Genetics and Forensic Toxicology, National Board of Forensic Medicine, Linköping, Sweden ; Clinical Pharmacology, Department of Health and Medical Sciences, Linköping University, Linköping, Sweden.
PLoS One. 2014 Jan 7;9(1):e84785. doi: 10.1371/journal.pone.0084785. eCollection 2014.
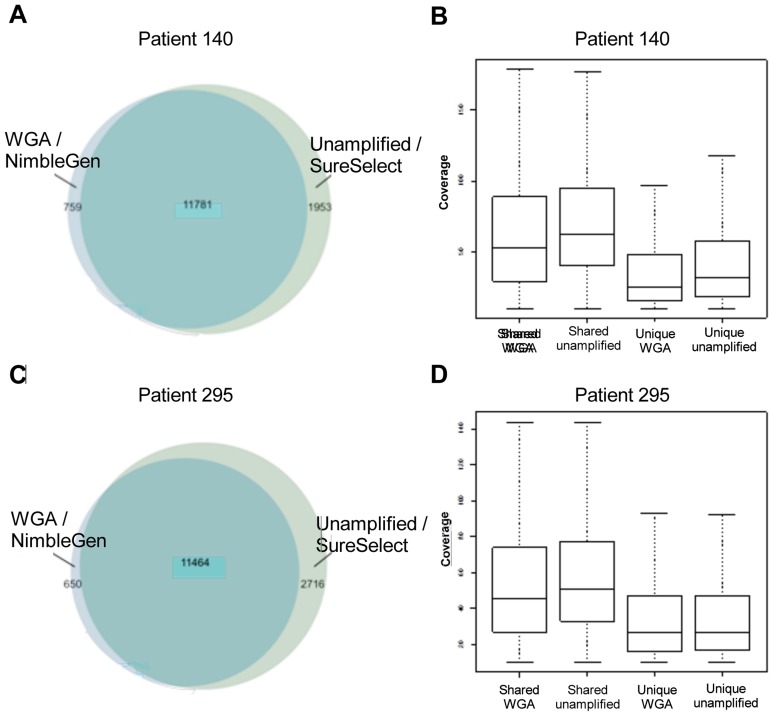
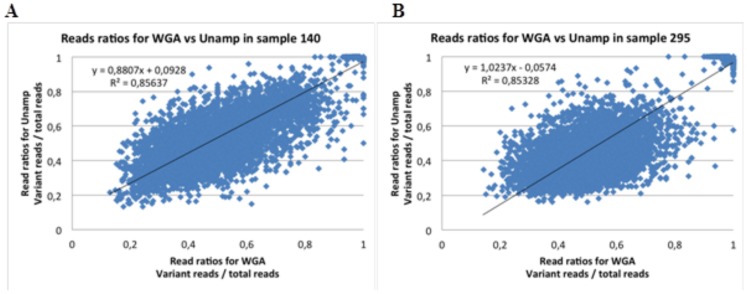
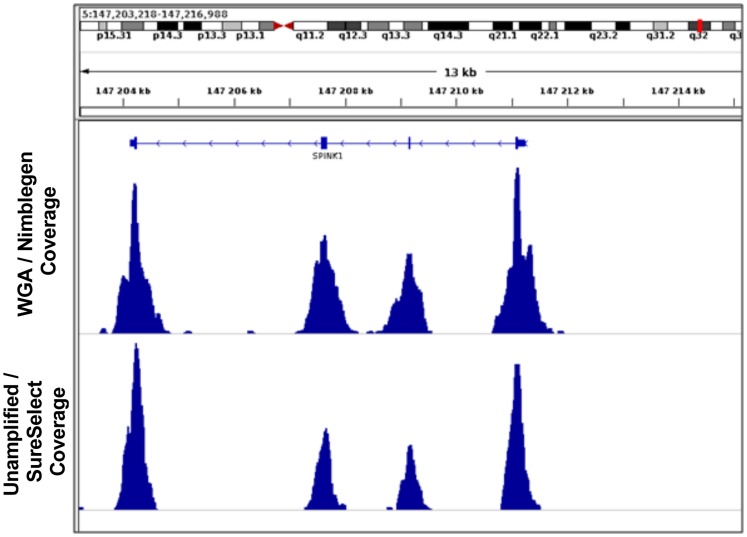
Exome sequence capture and massively parallel sequencing can be combined to achieve inexpensive and rapid global analyses of the functional sections of the genome. The difficulties of working with relatively small quantities of genetic material, as may be necessary when sharing tumor biopsies between collaborators for instance, can be overcome using whole genome amplification. However, the potential drawbacks of using a whole genome amplification technology based on random primers in combination with sequence capture followed by massively parallel sequencing have not yet been examined in detail, especially in the context of mutation discovery in tumor material. In this work, we compare mutations detected in sequence data for unamplified DNA, whole genome amplified DNA, and RNA originating from the same tumor tissue samples from 16 patients diagnosed with non-small cell lung cancer. The results obtained provide a comprehensive overview of the merits of these techniques for mutation analysis. We evaluated the identified genetic variants, and found that most (74%) of them were observed in both the amplified and the unamplified sequence data. Eighty-nine percent of the variations found by WGA were shared with unamplified DNA. We demonstrate a strategy for avoiding allelic bias by including RNA-sequencing information.
外显子组序列捕获和大规模并行测序可以结合起来,实现对基因组功能区域的廉价和快速的全球分析。在与合作者分享肿瘤活检等相对较小量的遗传物质时,可能会遇到工作上的困难,而使用全基因组扩增技术可以克服这些困难。然而,在肿瘤材料的突变发现方面,尚未详细研究基于随机引物的全基因组扩增技术与序列捕获和大规模并行测序相结合的潜在缺点。在这项工作中,我们比较了 16 名非小细胞肺癌患者的同一肿瘤组织样本的未经扩增的 DNA、全基因组扩增的 DNA 和 RNA 的序列数据中检测到的突变。得到的结果提供了这些技术在突变分析方面的优缺点的全面概述。我们评估了所鉴定的遗传变异,发现它们中的大多数(74%)在扩增和未扩增的序列数据中都被观察到。WGA 发现的 89%的变异与未扩增的 DNA 共享。我们通过包括 RNA 测序信息来展示避免等位基因偏倚的策略。