Duitama Jorge, Quintero Juan Camilo, Cruz Daniel Felipe, Quintero Constanza, Hubmann Georg, Foulquié-Moreno Maria R, Verstrepen Kevin J, Thevelein Johan M, Tohme Joe
Agrobiodiversity research area, International Center for Tropical Agriculture (CIAT), Km 17 Recta Cali- Palmira, A.A. 6713 Cali, Colombia, Laboratory of Molecular Cell Biology, Department of Biology, Institute of Botany and Microbiology, KU Leuven, Kasteelpark Arenberg 31, B-3001 Leuven-Heverlee, Flanders, Belgium, Department of Molecular Microbiology, VIB, Kasteelpark Arenberg 31, B-3001 Leuven-Heverlee, Flanders, Belgium, VIB Laboratory of Systems Biology, KU Leuven, Gaston Geenslaan 1, B-3001 Leuven-Heverlee, Flanders, Belgium and Laboratory for Genetics and Genomics, Centre of Microbial and Plant Genetics, KU Leuven, Gaston Geenslaan 1, B-3001 Leuven-Heverlee, Flanders, Belgium.
Nucleic Acids Res. 2014 Apr;42(6):e44. doi: 10.1093/nar/gkt1381. Epub 2014 Jan 11.
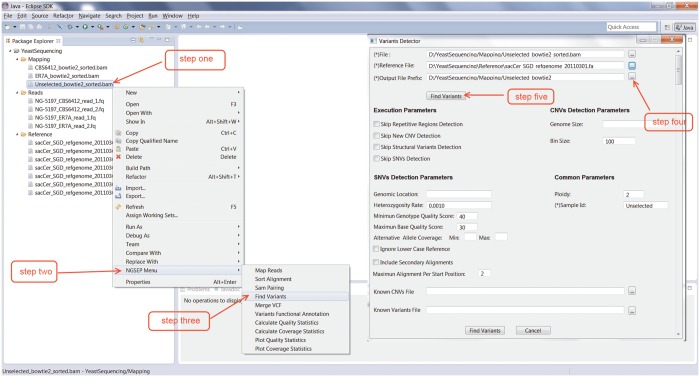
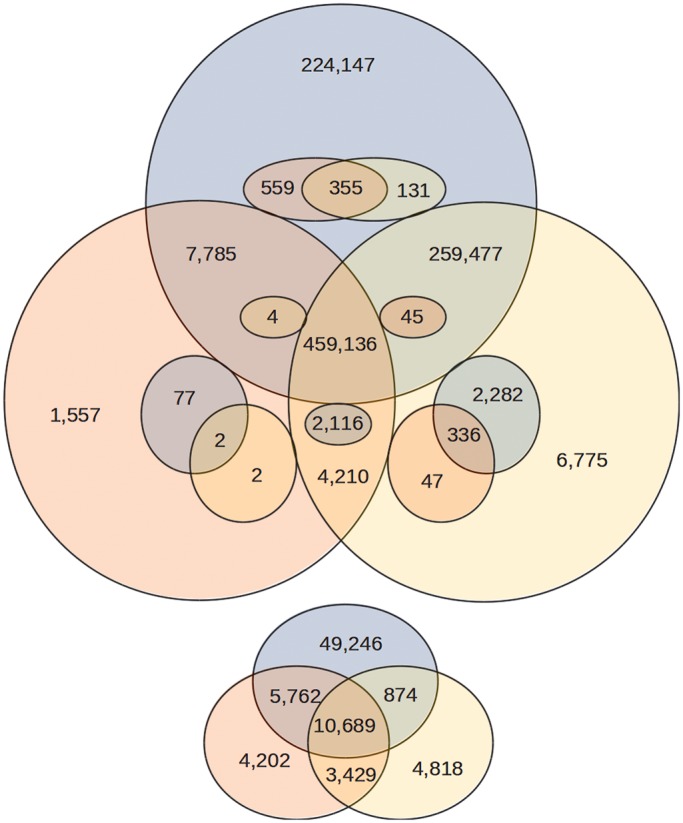
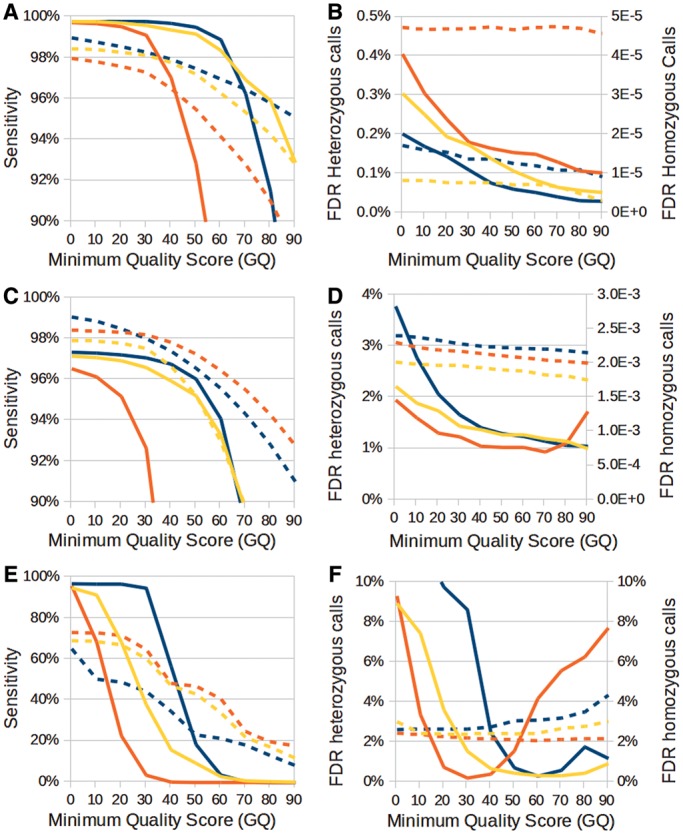
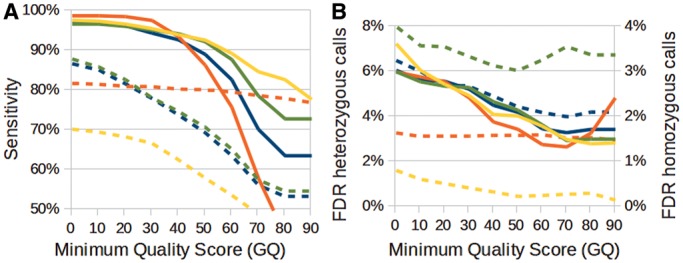
Recent advances in high-throughput sequencing (HTS) technologies and computing capacity have produced unprecedented amounts of genomic data that have unraveled the genetics of phenotypic variability in several species. However, operating and integrating current software tools for data analysis still require important investments in highly skilled personnel. Developing accurate, efficient and user-friendly software packages for HTS data analysis will lead to a more rapid discovery of genomic elements relevant to medical, agricultural and industrial applications. We therefore developed Next-Generation Sequencing Eclipse Plug-in (NGSEP), a new software tool for integrated, efficient and user-friendly detection of single nucleotide variants (SNVs), indels and copy number variants (CNVs). NGSEP includes modules for read alignment, sorting, merging, functional annotation of variants, filtering and quality statistics. Analysis of sequencing experiments in yeast, rice and human samples shows that NGSEP has superior accuracy and efficiency, compared with currently available packages for variants detection. We also show that only a comprehensive and accurate identification of repeat regions and CNVs allows researchers to properly separate SNVs from differences between copies of repeat elements. We expect that NGSEP will become a strong support tool to empower the analysis of sequencing data in a wide range of research projects on different species.
高通量测序(HTS)技术和计算能力的最新进展产生了前所未有的大量基因组数据,这些数据揭示了多个物种表型变异的遗传学机制。然而,操作和整合当前用于数据分析的软件工具仍需要在高技能人员方面进行大量投入。开发用于HTS数据分析的准确、高效且用户友好的软件包将有助于更快速地发现与医学、农业和工业应用相关的基因组元件。因此,我们开发了新一代测序Eclipse插件(NGSEP),这是一种用于集成、高效且用户友好地检测单核苷酸变异(SNV)、插入缺失(indel)和拷贝数变异(CNV)的新软件工具。NGSEP包括用于读段比对、排序、合并、变异功能注释、过滤和质量统计的模块。对酵母、水稻和人类样本测序实验的分析表明,与目前可用的变异检测软件包相比,NGSEP具有更高的准确性和效率。我们还表明,只有全面准确地识别重复区域和CNV,研究人员才能正确地将SNV与重复元件拷贝之间的差异区分开来。我们预计NGSEP将成为一个强大的支持工具,助力不同物种的广泛研究项目中的测序数据分析。