School of Animal and Veterinary Sciences, Charles Sturt University, Wagga Wagga, New South Wales, Australia ; Graham Centre for Agricultural Innovation (NSW Department of Primary Industries and Charles Sturt University), Wagga Wagga, New South Wales, Australia.
Institute of Marine and Antarctic Studies, University of Tasmania, Hobart, Tasmania, Australia.
PLoS One. 2014 Jan 8;9(1):e85370. doi: 10.1371/journal.pone.0085370. eCollection 2014.
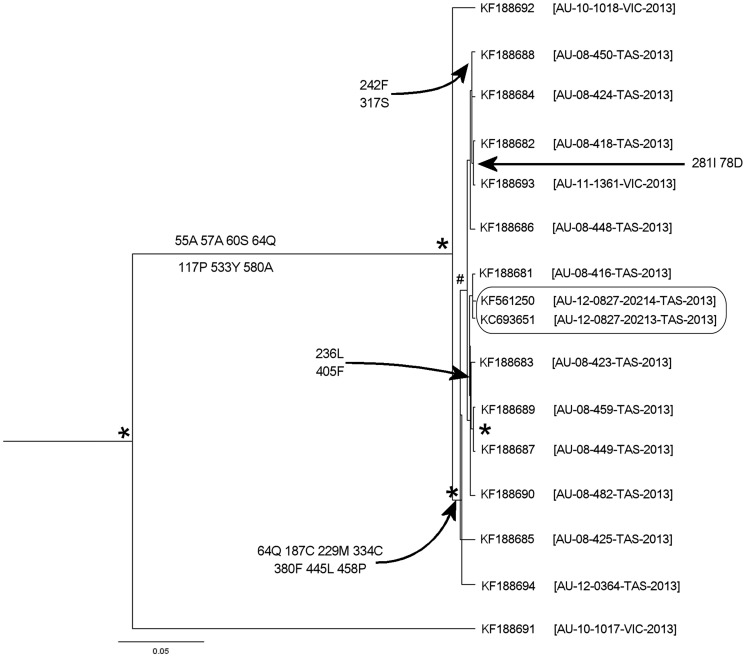
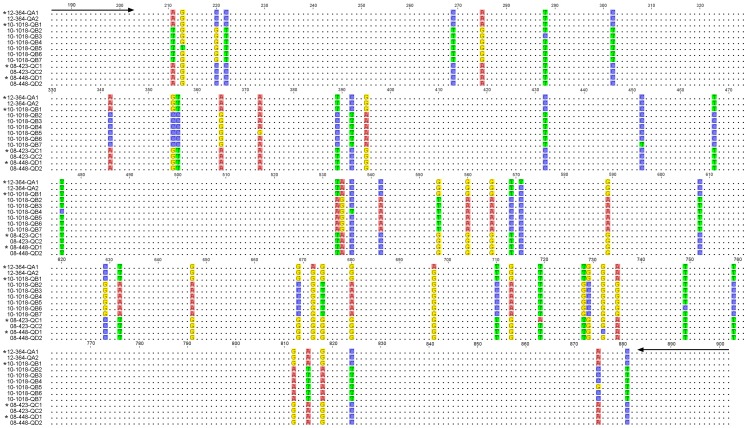
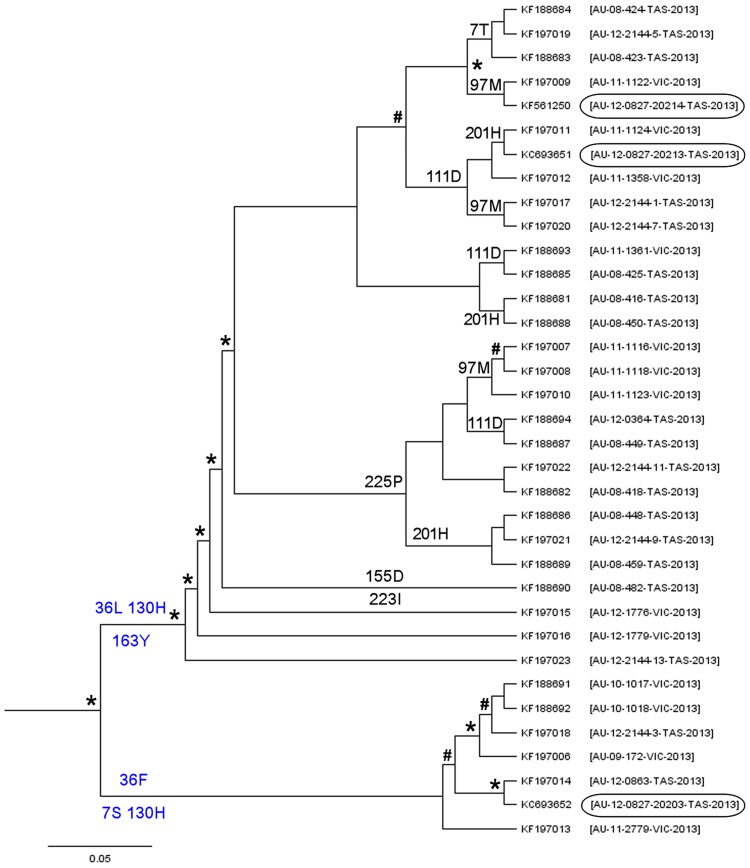
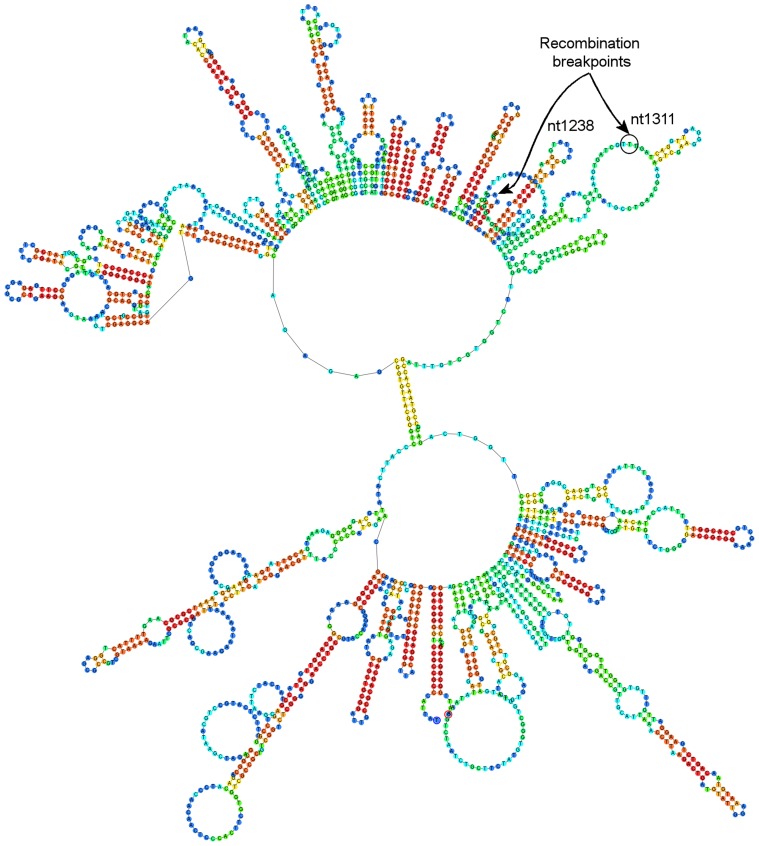
Quasispecies variants and recombination were studied longitudinally in an emergent outbreak of beak and feather disease virus (BFDV) infection in the orange-bellied parrot (Neophema chrysogaster). Detailed health monitoring and the small population size (<300 individuals) of this critically endangered bird provided an opportunity to longitudinally track viral replication and mutation events occurring in a circular, single-stranded DNA virus over a period of four years within a novel bottleneck population. Optimized PCR was used with different combinations of primers, primer walking, direct amplicon sequencing and sequencing of cloned amplicons to analyze BFDV genome variants. Analysis of complete viral genomes (n = 16) and Rep gene sequences (n = 35) revealed that the outbreak was associated with mutations in functionally important regions of the normally conserved Rep gene and immunogenic capsid (Cap) gene with a high evolutionary rate (3.41×10(-3) subs/site/year) approaching that for RNA viruses; simultaneously we observed significant evidence of recombination hotspots between two distinct progenitor genotypes within orange-bellied parrots indicating early cross-transmission of BFDV in the population. Multiple quasispecies variants were also demonstrated with at least 13 genotypic variants identified in four different individual birds, with one containing up to seven genetic variants. Preferential PCR amplification of variants was also detected. Our findings suggest that the high degree of genetic variation within the BFDV species as a whole is reflected in evolutionary dynamics within individually infected birds as quasispecies variation, particularly when BFDV jumps from one host species to another.
对喙羽病病毒(BFDV)感染暴发期间橙腹鹦鹉(Neophema chrysogaster)体内的准种变异和重组进行了纵向研究。对这种极度濒危鸟类进行详细的健康监测和对其小规模种群(<300 只)的研究为在一个新的瓶颈种群中,对一种环状单链 DNA 病毒在四年时间内发生的病毒复制和突变事件进行纵向追踪提供了机会。使用不同组合的引物、引物步移、直接扩增子测序和克隆扩增子测序对 BFDV 基因组变异进行了优化 PCR 分析。对完整病毒基因组(n = 16)和 Rep 基因序列(n = 35)的分析表明,该暴发与正常保守的 Rep 基因和免疫原性衣壳(Cap)基因功能重要区域的突变有关,其进化率很高(3.41×10(-3) 取代/位点/年),接近 RNA 病毒的进化率;同时,我们观察到橙腹鹦鹉内两个不同祖先生殖基因之间存在明显的重组热点,表明 BFDV 在该种群中的早期交叉传播。还证明了存在多种准种变异,在四只不同个体鸟类中发现了至少 13 种基因型变异,其中一种含有多达七种遗传变异。还检测到了变体的优先 PCR 扩增。我们的研究结果表明,BFDV 种内的高度遗传变异反映在个体感染鸟类内的进化动态中,表现为准种变异,尤其是当 BFDV 从一种宿主物种跳跃到另一种宿主物种时。