School of Biological and Population Health Sciences, College of Public Health and Human Sciences, Oregon State University, Corvallis, OR 97331, USA and Section of Biostatistics and Epidemiology, Department of Community and Family Medicine, Geisel School of Medicine at Dartmouth, Hanover, NH 03755, USA.
Bioinformatics. 2014 May 15;30(10):1431-9. doi: 10.1093/bioinformatics/btu029. Epub 2014 Jan 21.
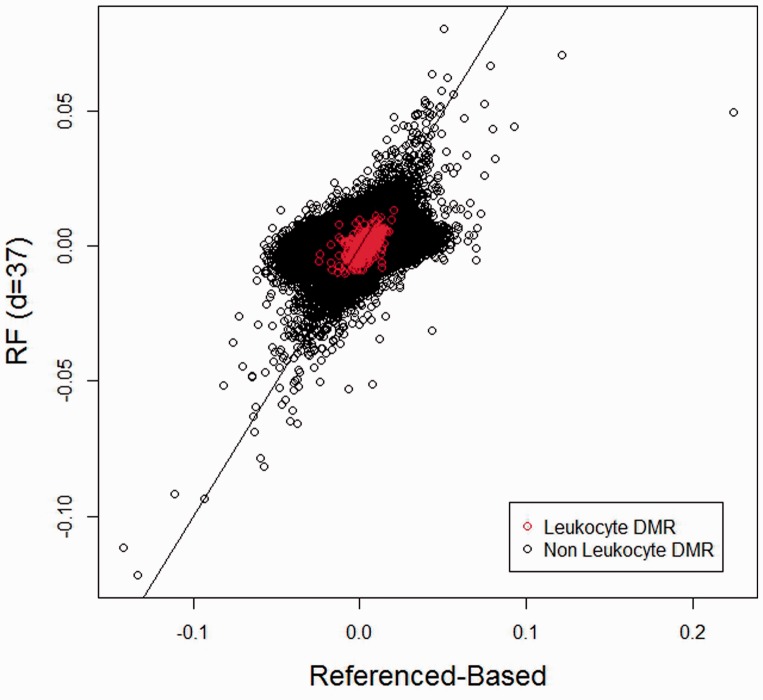
Recently there has been increasing interest in the effects of cell mixture on the measurement of DNA methylation, specifically the extent to which small perturbations in cell mixture proportions can register as changes in DNA methylation. A recently published set of statistical methods exploits this association to infer changes in cell mixture proportions, and these methods are presently being applied to adjust for cell mixture effect in the context of epigenome-wide association studies. However, these adjustments require the existence of reference datasets, which may be laborious or expensive to collect. For some tissues such as placenta, saliva, adipose or tumor tissue, the relevant underlying cell types may not be known.
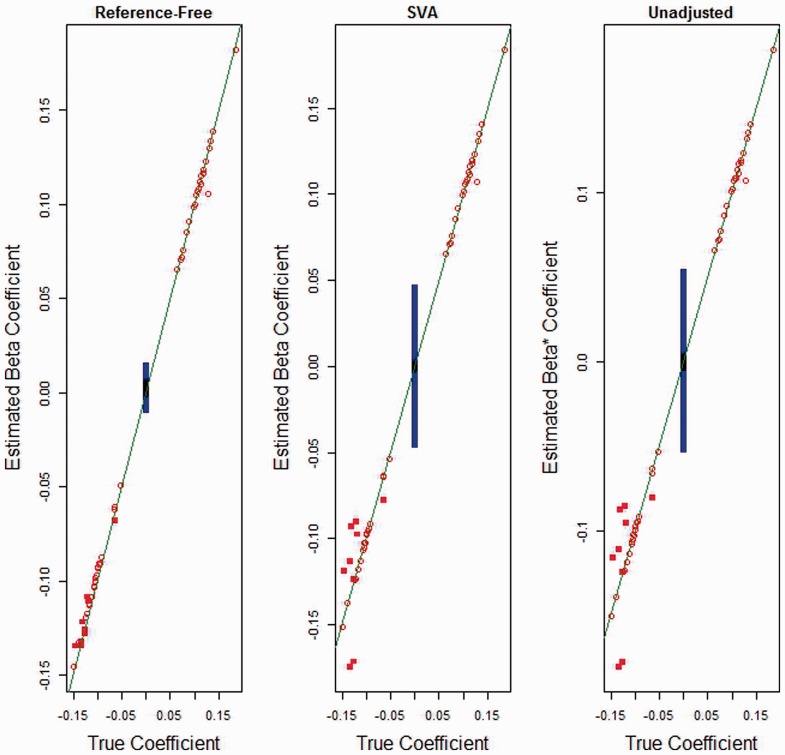
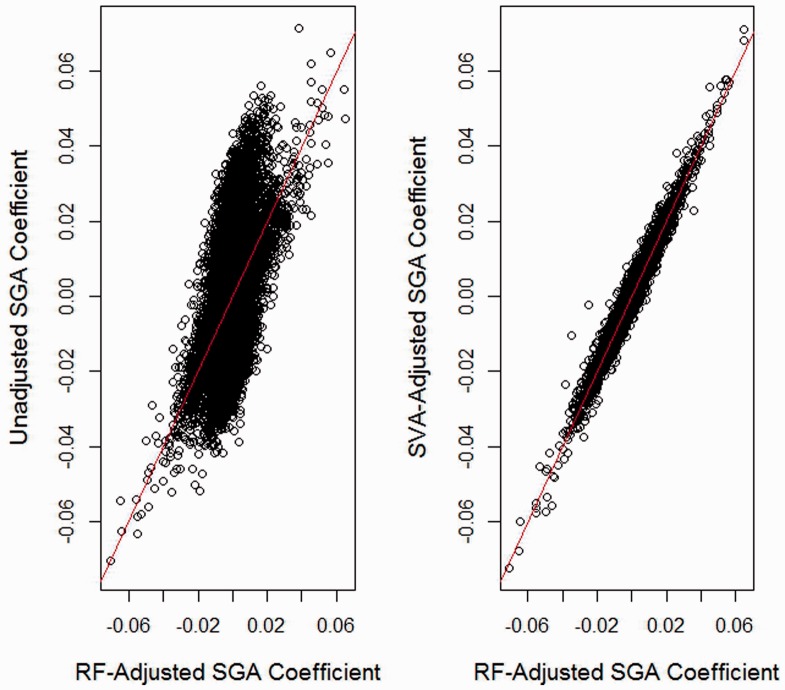
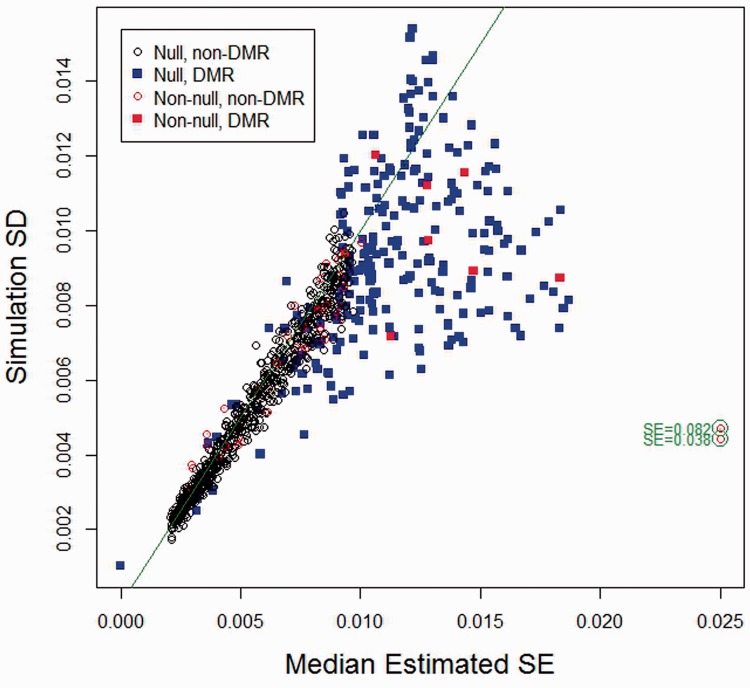
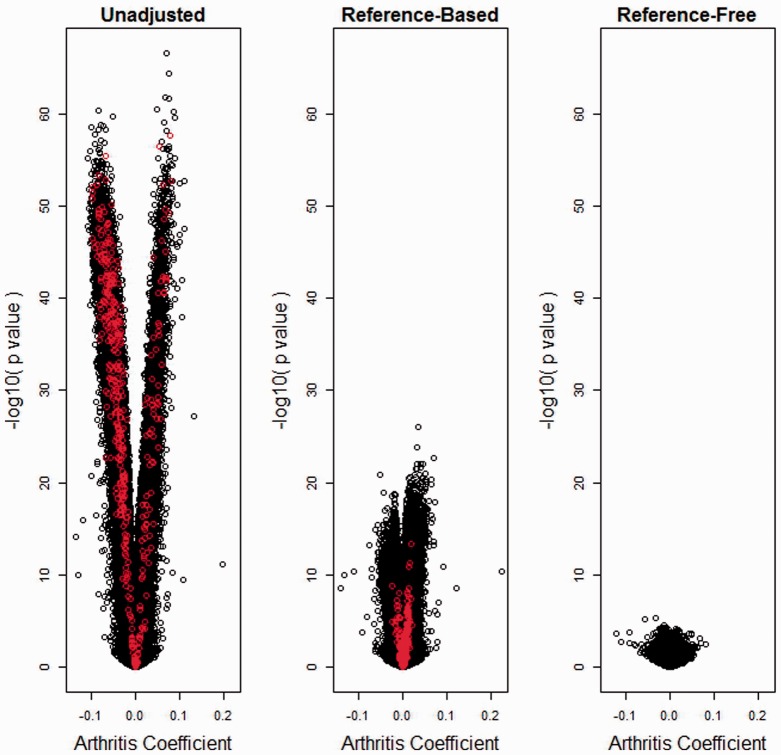
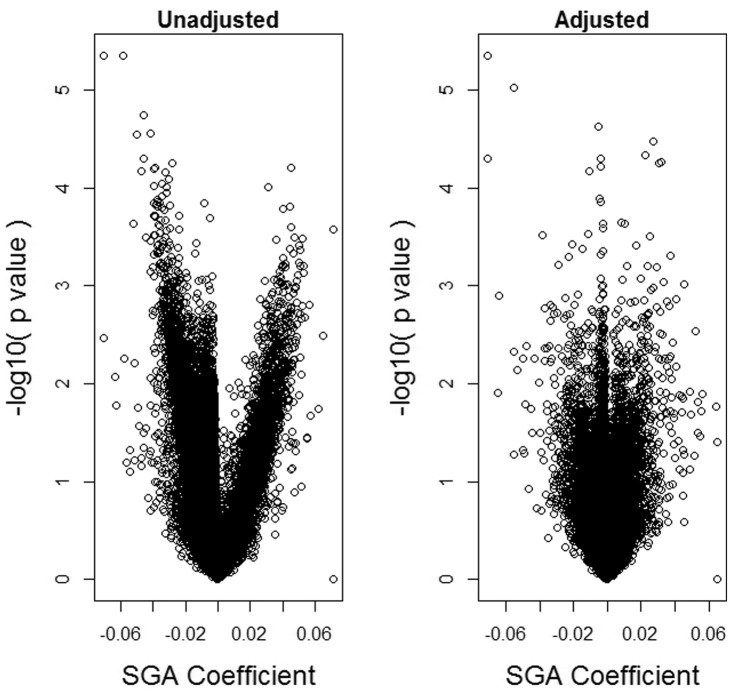
We propose a method for conducting epigenome-wide association studies analysis when a reference dataset is unavailable, including a bootstrap method for estimating standard errors. We demonstrate via simulation study and several real data analyses that our proposed method can perform as well as or better than methods that make explicit use of reference datasets. In particular, it may adjust for detailed cell type differences that may be unavailable even in existing reference datasets.
Software is available in the R package RefFreeEWAS. Data for three of four examples were obtained from Gene Expression Omnibus (GEO), accession numbers GSE37008, GSE42861 and GSE30601, while reference data were obtained from GEO accession number GSE39981.
andres.houseman@oregonstate.edu
Supplementary data are available at Bioinformatics online.
最近,人们对细胞混合物对 DNA 甲基化测量的影响越来越感兴趣,特别是细胞混合物比例的微小扰动在多大程度上可以记录为 DNA 甲基化的变化。最近发表的一组统计方法利用这种关联来推断细胞混合物比例的变化,目前这些方法正被应用于调整表观基因组全关联研究中的细胞混合物效应。然而,这些调整需要存在参考数据集,而这些数据集可能难以收集或昂贵。对于某些组织,如胎盘、唾液、脂肪或肿瘤组织,相关的潜在细胞类型可能未知。
我们提出了一种在没有参考数据集的情况下进行全基因组关联研究分析的方法,包括一种用于估计标准误差的自举方法。通过模拟研究和几个真实数据分析,我们证明了我们提出的方法可以与明确使用参考数据集的方法一样或更好地进行分析。特别是,它可以调整即使在现有的参考数据集中也可能无法获得的详细细胞类型差异。
软件可在 R 包 RefFreeEWAS 中使用。四个示例中的三个数据来自基因表达综合数据库(GEO),注册号为 GSE37008、GSE42861 和 GSE30601,而参考数据则来自 GEO 注册号 GSE39981。
andres.houseman@oregonstate.edu
补充数据可在“生物信息学在线”上获得。