Department of Integrative Biology, University of California, Berkeley, California, United States of America.
Department of Plant and Microbial Biology, University of California, Berkeley, California, United States of America.
PLoS One. 2014 Jan 21;9(1):e85103. doi: 10.1371/journal.pone.0085103. eCollection 2014.
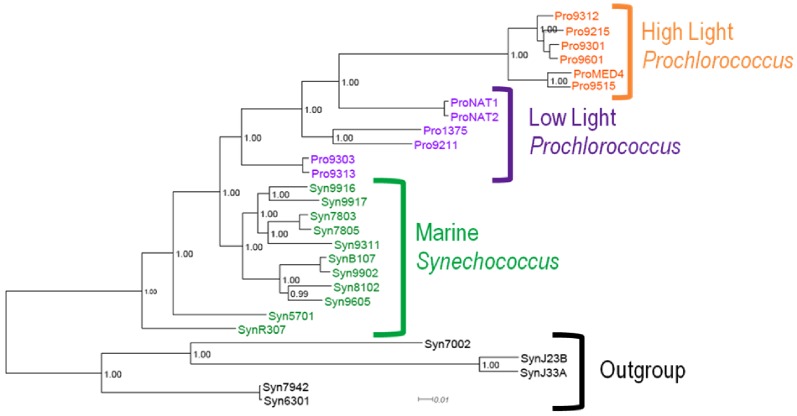
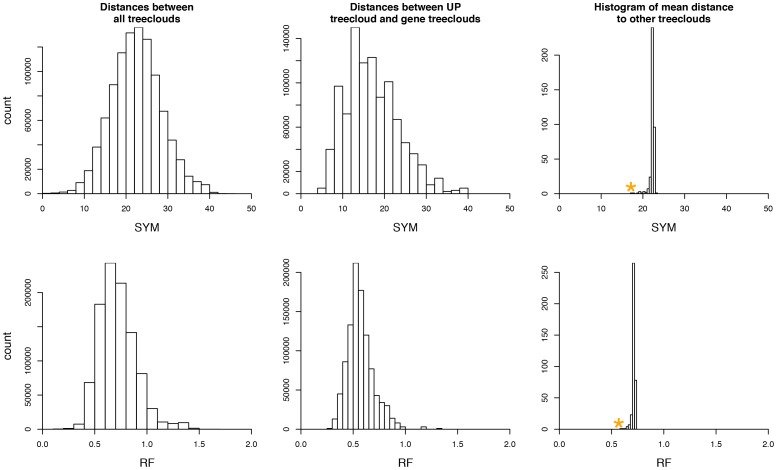
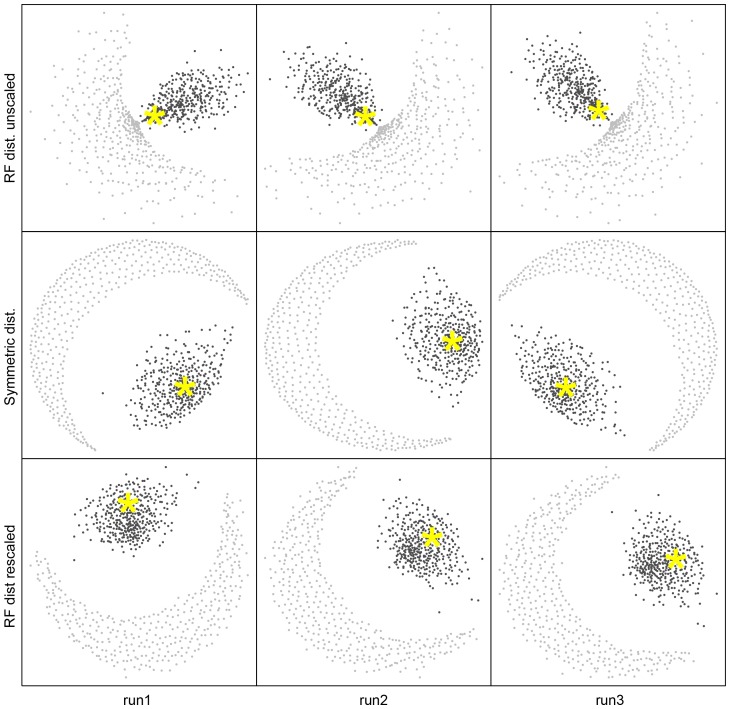
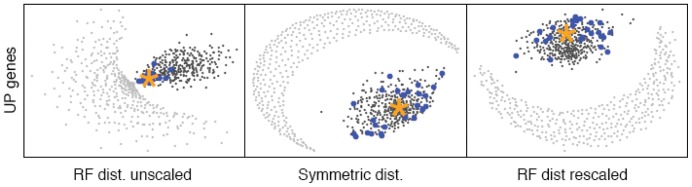
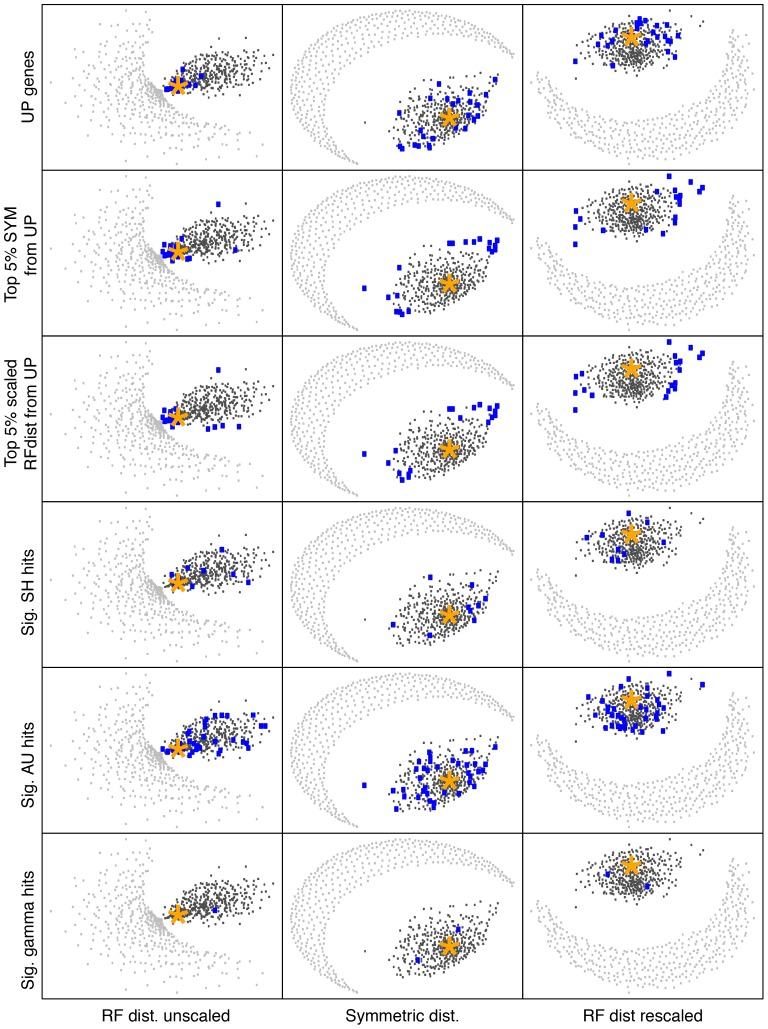
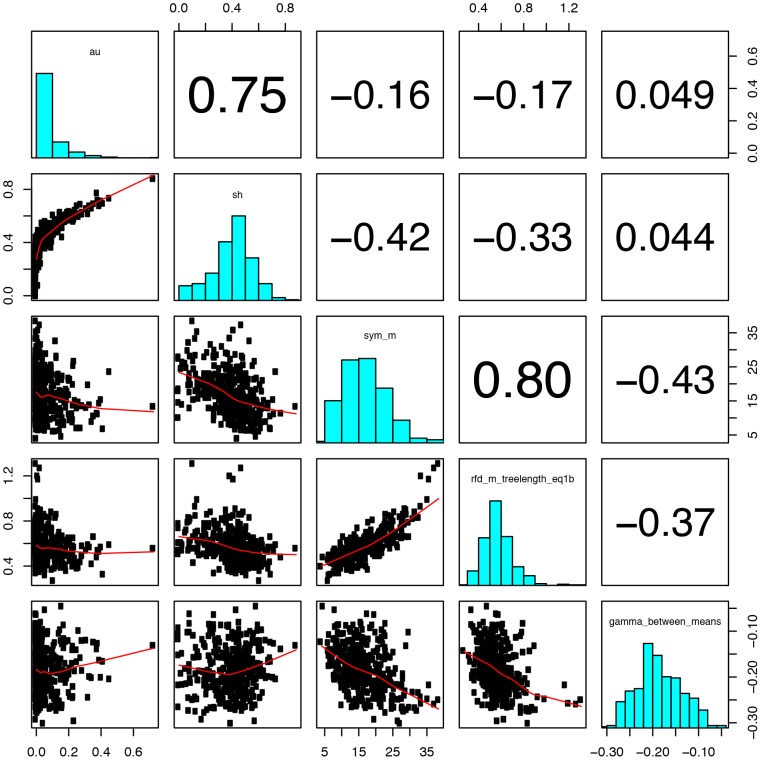
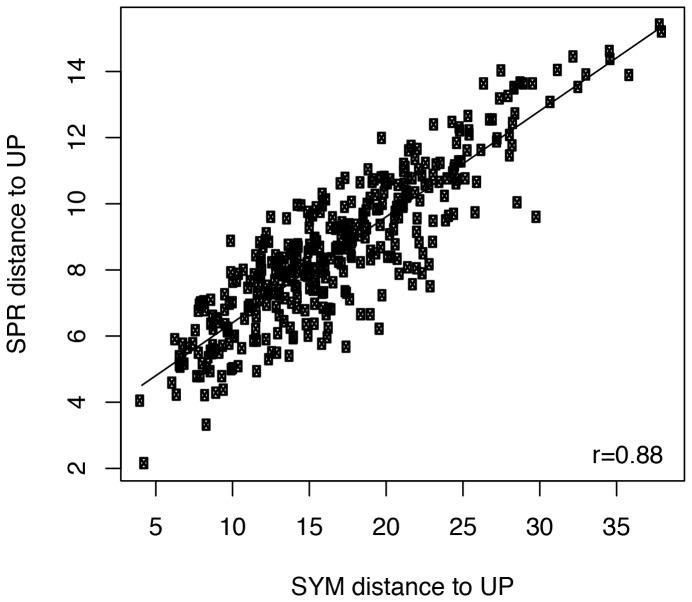
It is often suggested that horizontal gene transfer is so ubiquitous in microbes that the concept of a phylogenetic tree representing the pattern of vertical inheritance is oversimplified or even positively misleading. "Universal proteins" have been used to infer the organismal phylogeny, but have been criticized as being only the "tree of one percent." Currently, few options exist for those wishing to rigorously assess how well a universal protein phylogeny, based on a relative handful of well-conserved genes, represents the phylogenetic histories of hundreds of genes. Here, we address this problem by proposing a visualization method and a statistical test within a Bayesian framework. We use the genomes of marine cyanobacteria, a group thought to exhibit substantial amounts of HGT, as a test case. We take 379 orthologous gene families from 28 cyanobacteria genomes and estimate the Bayesian posterior distributions of trees - a "treecloud" - for each, as well as for a concatenated dataset based on putative "universal proteins." We then calculate the average distance between trees within and between all treeclouds on various metrics and visualize this high-dimensional space with non-metric multidimensional scaling (NMMDS). We show that the tree space is strongly clustered and that the universal protein treecloud is statistically significantly closer to the center of this tree space than any individual gene treecloud. We apply several commonly-used tests for incongruence/HGT and show that they agree HGT is rare in this dataset, but make different choices about which genes were subject to HGT. Our results show that the question of the representativeness of the "tree of one percent" is a quantitative empirical question, and that the phylogenetic central tendency is a meaningful observation even if many individual genes disagree due to the various sources of incongruence.
人们常说,水平基因转移在微生物中如此普遍,以至于代表垂直遗传模式的系统发育树的概念过于简单化,甚至具有误导性。“普遍蛋白”被用来推断生物系统发育,但也被批评为只是“百分之一的树”。目前,对于那些希望严格评估基于少数几个高度保守基因的普遍蛋白系统发育树在多大程度上代表数百个基因的系统发育历史的人来说,选择很少。在这里,我们通过在贝叶斯框架内提出一种可视化方法和一种统计检验来解决这个问题。我们以海洋蓝藻作为一个测试案例,这是一个被认为存在大量 HGT 的群体。我们从 28 种蓝藻基因组中选取了 379 个直系同源基因家族,并为每个基因家族以及基于假定的“普遍蛋白”的串联数据集估计了贝叶斯后验树分布——“树云”。然后,我们计算了在各种度量标准下所有树云中的树内和树间的平均距离,并使用非度量多维缩放 (NMMDS) 可视化这个高维空间。我们表明,树空间强烈聚类,并且普遍蛋白树云在统计上明显更接近该树空间的中心,而不是任何单个基因树云。我们应用了几种常用的用于不一致性/水平基因转移的检验,并表明它们一致认为在这个数据集水平基因转移很少见,但对于哪些基因受到水平基因转移的影响有不同的选择。我们的结果表明,“百分之一的树”的代表性问题是一个定量的经验问题,即使由于各种不一致性的来源,许多个别基因存在分歧,系统发育的中心趋势仍然是一个有意义的观察结果。