Sackler Institute for Comparative Genomics, American Museum of Natural History, USA.
Genome Biol Evol. 2012;4(1):30-43. doi: 10.1093/gbe/evr121. Epub 2011 Nov 16.
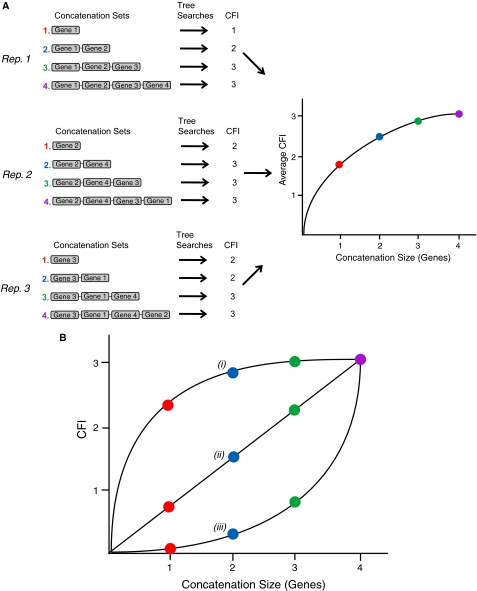
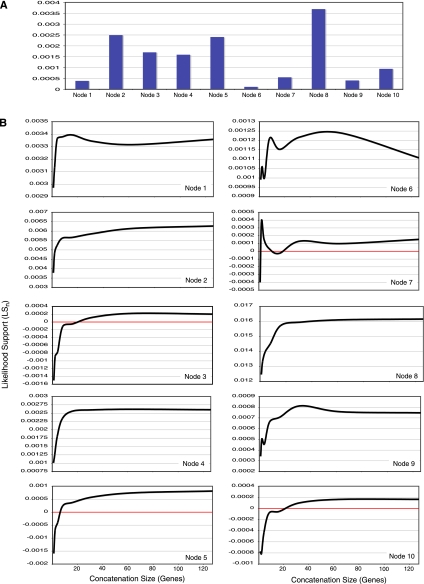
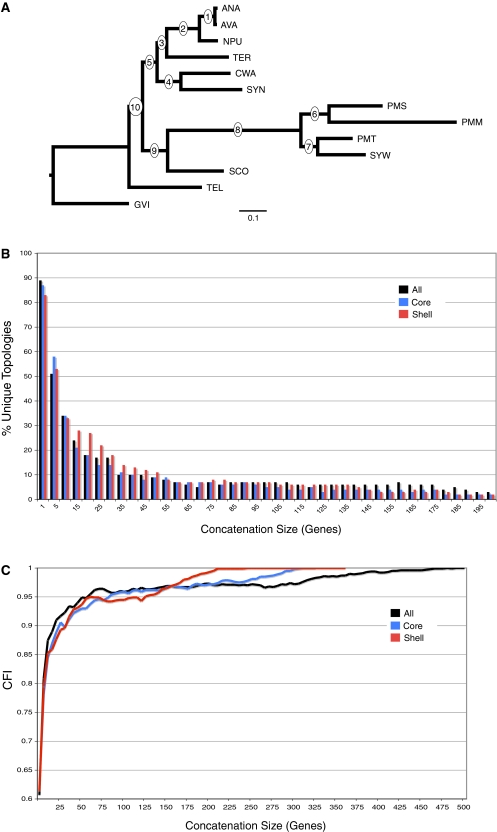
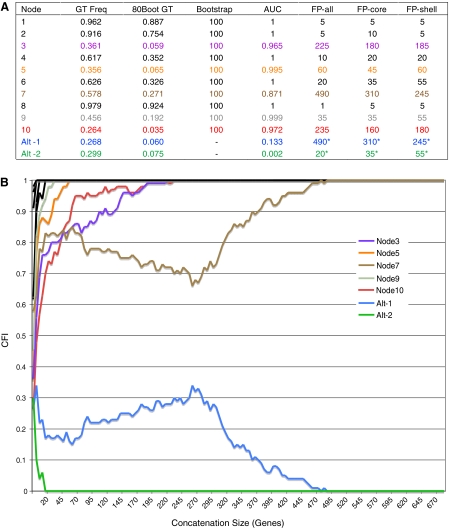
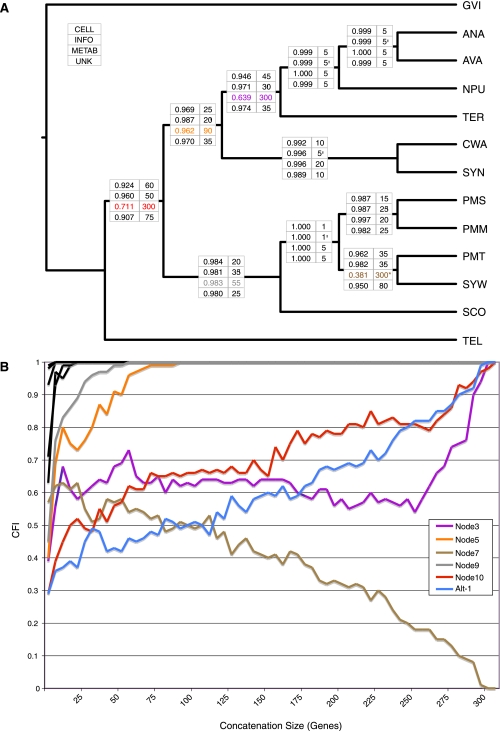
Recent whole-genome approaches to microbial phylogeny have emphasized partitioning genes into functional classes, often focusing on differences between a stable core of genes and a variable shell. To rigorously address the effects of partitioning and combining genes in genome-level analyses, we developed a novel technique called Random Addition Concatenation Analysis (RADICAL). RADICAL operates by sequentially concatenating randomly chosen gene partitions starting with a single-gene partition and ending with the entire genomic data set. A phylogenetic tree is built for every successive addition, and the entire process is repeated creating multiple random concatenation paths. The result is a library of trees representing a large variety of differently sized random gene partitions. This library can then be mined to identify unique topologies, assess overall agreement, and measure support for different trees. To evaluate RADICAL, we used 682 orthologous genes across 13 cyanobacterial genomes. Despite previous assertions of substantial differences between a core and a shell set of genes for this data set, RADICAL reveals the two partitions contain congruent phylogenetic signal. Substantial disagreement within the data set is limited to a few nodes and genes involved in metabolism, a functional group that is distributed evenly between the core and the shell partitions. We highlight numerous examples where RADICAL reveals aspects of phylogenetic behavior not evident by examining individual gene trees or a "'total evidence" tree. Our method also demonstrates that most emergent phylogenetic signal appears early in the concatenation process. The software is freely available at http://desalle.amnh.org.
最近的微生物系统发育全基因组方法强调将基因划分为功能类别,通常侧重于稳定核心基因和可变外壳基因之间的差异。为了严格解决在基因组水平分析中划分和组合基因的影响,我们开发了一种称为随机添加连接分析(RADICAL)的新技术。RADICAL 通过顺序连接随机选择的基因分区来操作,从单个基因分区开始,以整个基因组数据集结束。为每个连续的添加构建一个系统发育树,并且重复整个过程以创建多个随机连接路径。结果是一个代表各种不同大小的随机基因分区的树库。然后可以对该库进行挖掘,以识别独特的拓扑结构,评估总体一致性,并衡量不同树的支持度。为了评估 RADICAL,我们使用了 13 个蓝藻基因组中 682 个直系同源基因。尽管先前断言该数据集的核心和外壳基因之间存在很大差异,但 RADICAL 显示这两个分区包含一致的系统发育信号。数据集内的大量分歧仅限于少数节点和参与代谢的基因,代谢是核心和外壳分区之间均匀分布的功能组。我们强调了许多例子,其中 RADICAL 揭示了通过检查单个基因树或“总证据”树不明显的系统发育行为的方面。我们的方法还表明,大多数新兴的系统发育信号似乎出现在连接过程的早期。该软件可在 http://desalle.amnh.org 免费获得。