Eggeling Ralf, Gohr André, Keilwagen Jens, Mohr Michaela, Posch Stefan, Smith Andrew D, Grosse Ivo
Institute of Computer Science, Martin Luther University Halle-Wittenberg, Halle/Saale, Germany.
Institute for Biosafety in Plant Biotechnology, Julius Kühn-Institut (JKI) - Federal Research Centre for Cultivated Plants, Quedlinburg, Germany ; Department of Genebank, Leibniz Institute of Plant Genetics and Crop Plant Research (IPK), Seeland OT Gatersleben, Germany.
PLoS One. 2014 Jan 22;9(1):e85629. doi: 10.1371/journal.pone.0085629. eCollection 2014.
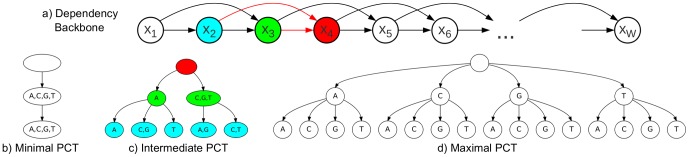
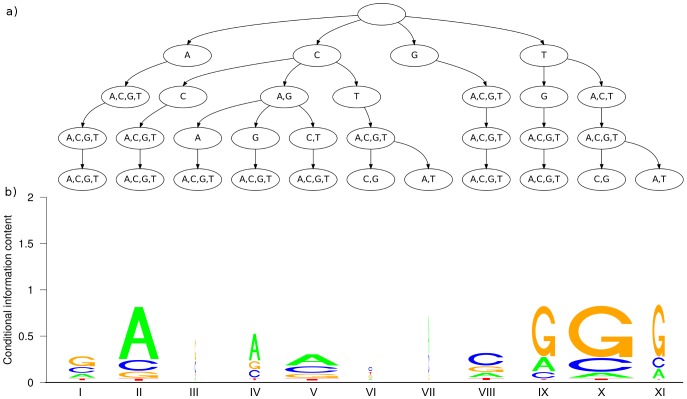
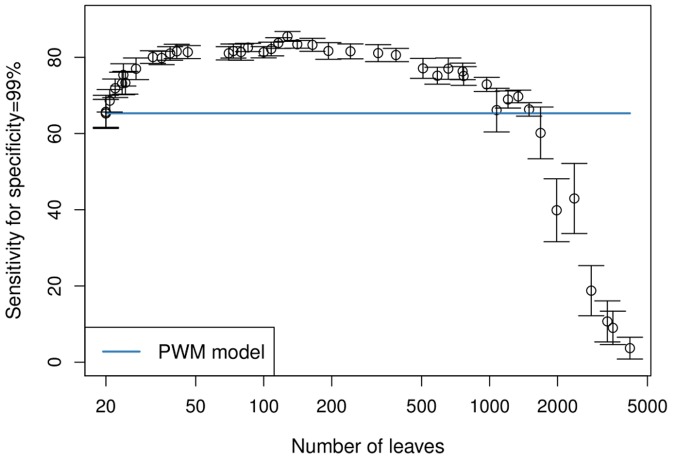
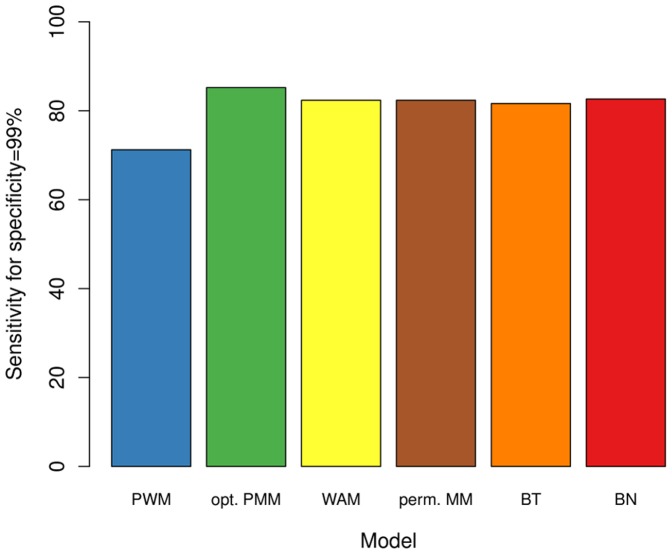
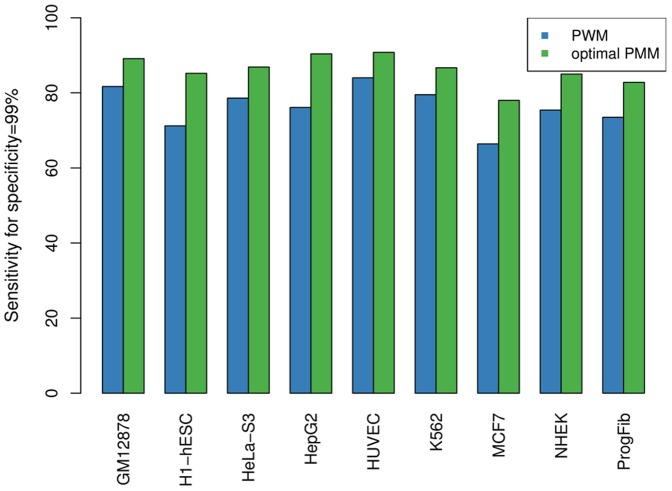
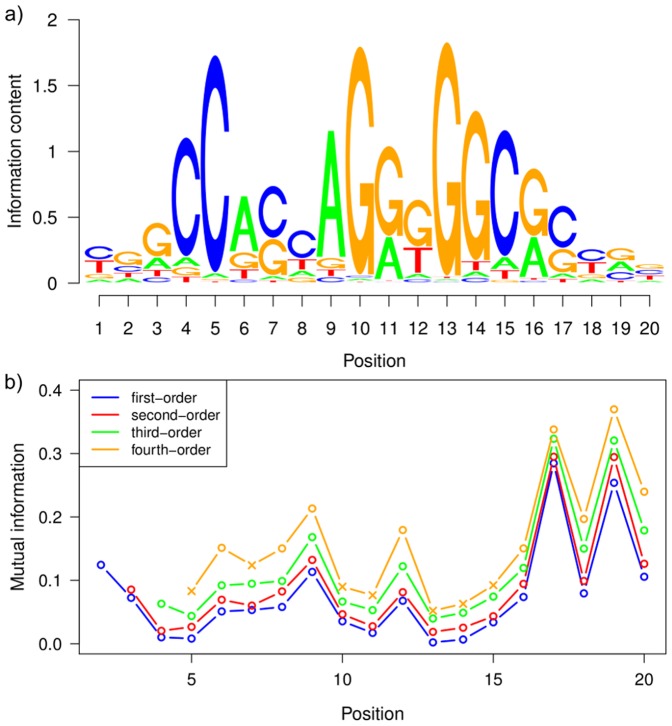
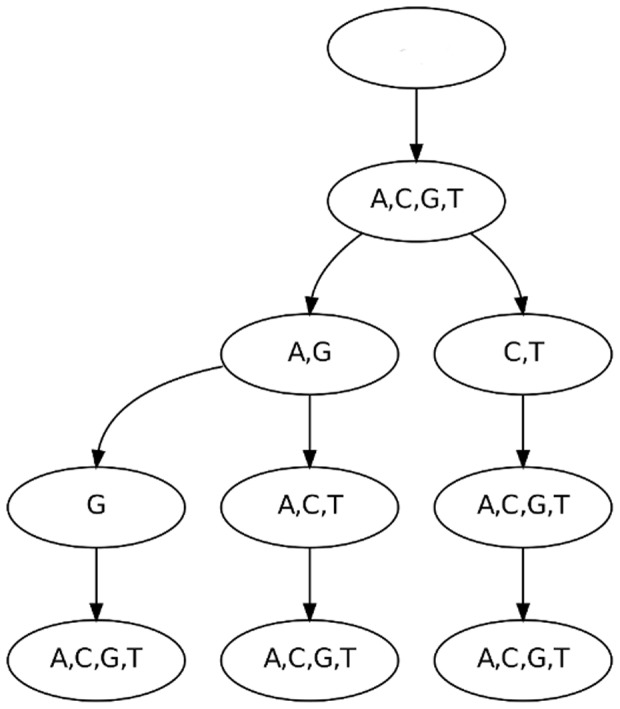
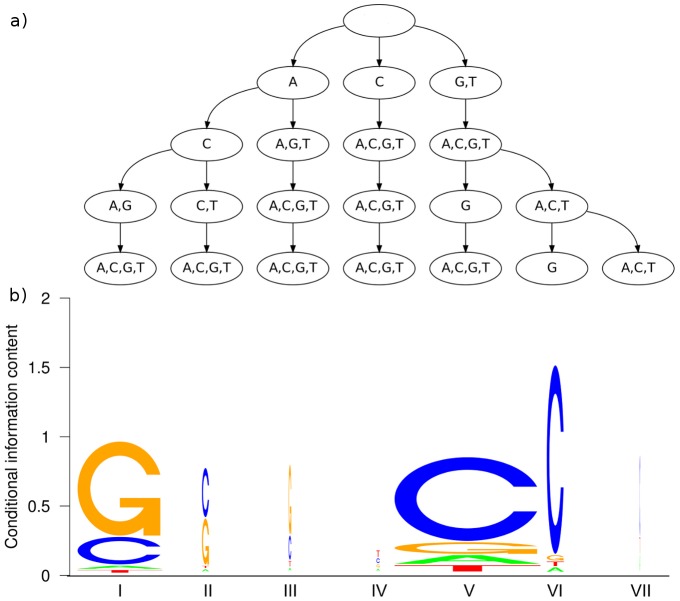
The binding affinity of DNA-binding proteins such as transcription factors is mainly determined by the base composition of the corresponding binding site on the DNA strand. Most proteins do not bind only a single sequence, but rather a set of sequences, which may be modeled by a sequence motif. Algorithms for de novo motif discovery differ in their promoter models, learning approaches, and other aspects, but typically use the statistically simple position weight matrix model for the motif, which assumes statistical independence among all nucleotides. However, there is no clear justification for that assumption, leading to an ongoing debate about the importance of modeling dependencies between nucleotides within binding sites. In the past, modeling statistical dependencies within binding sites has been hampered by the problem of limited data. With the rise of high-throughput technologies such as ChIP-seq, this situation has now changed, making it possible to make use of statistical dependencies effectively. In this work, we investigate the presence of statistical dependencies in binding sites of the human enhancer-blocking insulator protein CTCF by using the recently developed model class of inhomogeneous parsimonious Markov models, which is capable of modeling complex dependencies while avoiding overfitting. These findings lead to a more detailed characterization of the CTCF binding motif, which is only poorly represented by independent nucleotide frequencies at several positions, predominantly at the 3' end.
诸如转录因子等DNA结合蛋白的结合亲和力主要由DNA链上相应结合位点的碱基组成决定。大多数蛋白质并非只结合单一序列,而是结合一组序列,这组序列可用序列基序来建模。从头基序发现算法在其启动子模型、学习方法及其他方面存在差异,但通常使用统计上简单的位置权重矩阵模型来表示基序,该模型假定所有核苷酸之间具有统计独立性。然而,这种假设并无明确的依据,这引发了关于对结合位点内核苷酸之间的相关性进行建模的重要性的持续争论。过去,由于数据有限的问题,对结合位点内的统计相关性进行建模受到了阻碍。随着ChIP-seq等高通量技术的兴起,这种情况现在已经改变,使得有效利用统计相关性成为可能。在这项工作中,我们通过使用最近开发的非齐次简约马尔可夫模型类别来研究人类增强子阻断绝缘子蛋白CTCF结合位点中统计相关性的存在,该模型能够在避免过度拟合的同时对复杂的相关性进行建模。这些发现导致对CTCF结合基序有了更详细的表征,在几个位置,主要是在3'端,独立核苷酸频率对其的表征较差。