Institute of Genetic Medicine, Newcastle University, International Centre for Life, Central Parkway, Newcastle upon Tyne, United Kingdom.
Genet Epidemiol. 2014 Apr;38(3):173-90. doi: 10.1002/gepi.21792. Epub 2014 Feb 17.
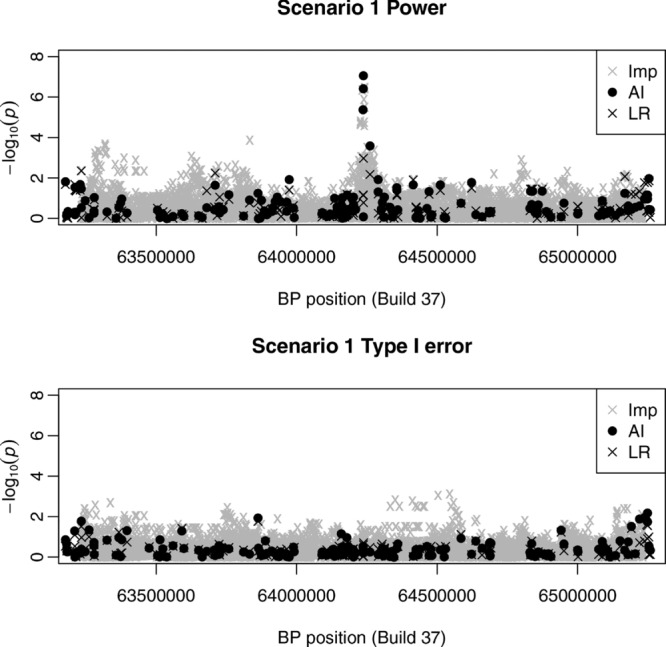
Genome-wide association studies allow detection of non-genotyped disease-causing variants through testing of nearby genotyped SNPs. This approach may fail when there are no genotyped SNPs in strong LD with the causal variant. Several genotyped SNPs in weak LD with the causal variant may, however, considered together, provide equivalent information. This observation motivates popular but computationally intensive approaches based on imputation or haplotyping. Here we present a new method and accompanying software designed for this scenario. Our approach proceeds by selecting, for each genotyped "anchor" SNP, a nearby genotyped "partner" SNP, chosen via a specific algorithm we have developed. These two SNPs are used as predictors in linear or logistic regression analysis to generate a final significance test. In simulations, our method captures much of the signal captured by imputation, while taking a fraction of the time and disc space, and generating a smaller number of false-positives. We apply our method to a case/control study of severe malaria genotyped using the Affymetrix 500K array. Previous analysis showed that fine-scale sequencing of a Gambian reference panel in the region of the known causal locus, followed by imputation, increased the signal of association to genome-wide significance levels. Our method also increases the signal of association from P ≈ 2 × 10⁻⁶ to P ≈ 6 × 10⁻¹¹. Our method thus, in some cases, eliminates the need for more complex methods such as sequencing and imputation, and provides a useful additional test that may be used to identify genetic regions of interest.
全基因组关联研究允许通过测试附近的基因分型 SNP 来检测未基因分型的致病变体。当与因果变体强连锁的基因分型 SNP 不存在时,这种方法可能会失败。然而,与因果变体弱连锁的几个基因分型 SNP 可以一起考虑,提供等效的信息。这一观察结果激发了基于推断或单体型的流行但计算密集型方法。在这里,我们提出了一种新的方法和相应的软件,专门用于这种情况。我们的方法通过为每个基因分型的“锚”SNP 选择附近的基因分型“伙伴”SNP 来进行,这些 SNP 是通过我们开发的特定算法选择的。这两个 SNP 被用作线性或逻辑回归分析的预测因子,以生成最终的显著性检验。在模拟中,我们的方法捕获了推断所捕获的大部分信号,同时占用了一小部分时间和磁盘空间,并产生了较少的假阳性。我们将我们的方法应用于使用 Affymetrix 500K 阵列进行严重疟疾基因分型的病例对照研究。先前的分析表明,在已知因果基因座区域对冈比亚参考面板进行精细测序,然后进行推断,可以增加关联信号达到全基因组显著性水平。我们的方法还将关联信号从 P ≈ 2 × 10⁻⁶增加到 P ≈ 6 × 10⁻¹¹。因此,在某些情况下,我们的方法消除了对更复杂方法(如测序和推断)的需求,并提供了一个有用的附加测试,可用于识别感兴趣的遗传区域。