BMC Genomics. 2013;14 Suppl 8(Suppl 8):S2. doi: 10.1186/1471-2164-14-S8-S2. Epub 2013 Dec 9.
RNAseq technology is replacing microarray technology as the tool of choice for gene expression profiling. While providing much richer data than microarray, analysis of RNAseq data has been much more challenging. To date, there has not been a consensus on the best approach for conducting robust RNAseq analysis.
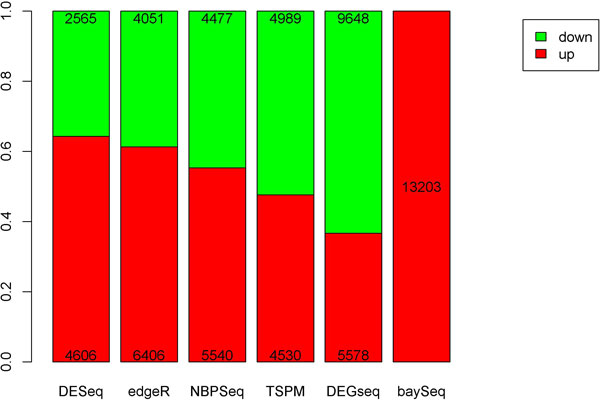
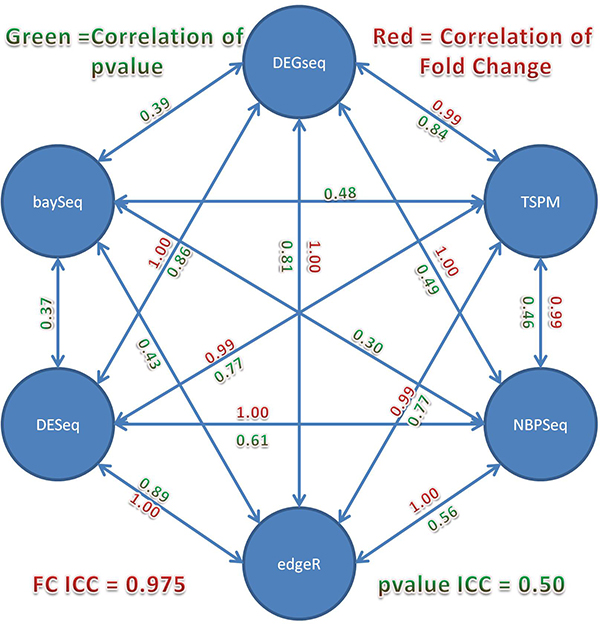
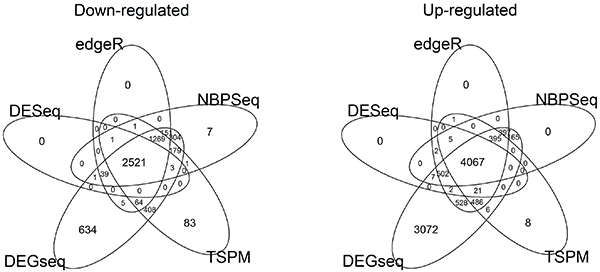
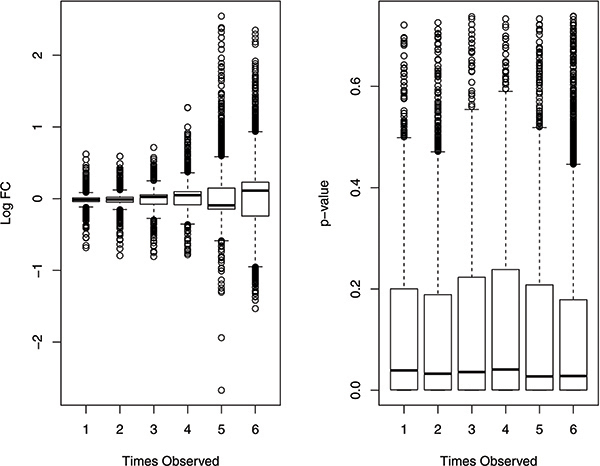
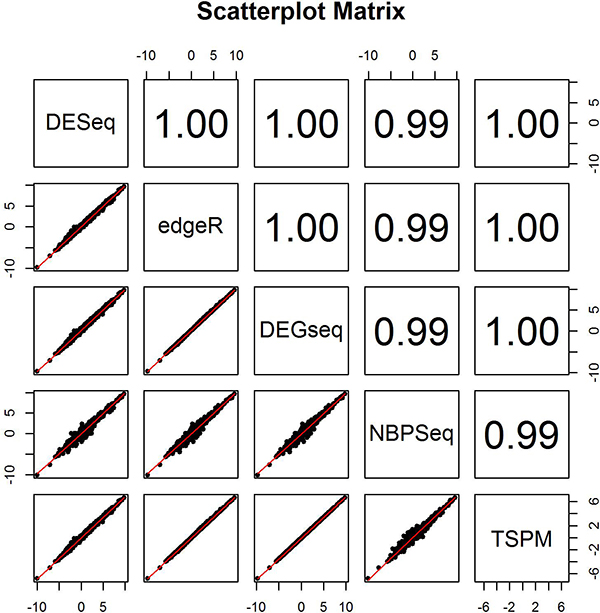
In this study, we designed a thorough experiment to evaluate six read count-based RNAseq analysis methods (DESeq, DEGseq, edgeR, NBPSeq, TSPM and baySeq) using both real and simulated data. We found the six methods produce similar fold changes and reasonable overlapping of differentially expressed genes based on p-values. However, all six methods suffer from over-sensitivity.
Based on the evaluation of runtime using real data and area under the receiver operating characteristic curve (AUC-ROC) using simulated data, we found that edgeR achieves a better balance between speed and accuracy than the other methods.
RNAseq 技术正在取代微阵列技术,成为基因表达谱分析的首选工具。虽然 RNAseq 技术比微阵列技术提供了更丰富的数据,但对其数据的分析也更具挑战性。迄今为止,对于进行稳健的 RNAseq 分析,还没有达成共识的最佳方法。
在这项研究中,我们设计了一个全面的实验,使用真实数据和模拟数据来评估六种基于读取计数的 RNAseq 分析方法(DESeq、DEGseq、edgeR、NBPSeq、TSPM 和 baySeq)。我们发现,基于 p 值,这六种方法产生的倍数变化和差异表达基因的合理重叠相似。然而,所有六种方法都存在过度敏感的问题。
基于使用真实数据评估运行时间和使用模拟数据评估接收者操作特征曲线下的面积(AUC-ROC),我们发现 edgeR 在速度和准确性之间取得了比其他方法更好的平衡。