Evolutionary Ecology and Genetics, Zoological Institute, CAU Kiel, Am Botanischen Garten 9, 24118, Kiel, Germany.
Institute for Clinical Molecular Biology, CAU Kiel, Am Botanischen Garten 11, 24118, Kiel, Germany.
BMC Genomics. 2019 May 10;20(1):364. doi: 10.1186/s12864-019-5686-1.
Data normalization and identification of significant differential expression represent crucial steps in RNA-Seq analysis. Many available tools rely on assumptions that are often not met by real data, including the common assumption of symmetrical distribution of up- and down-regulated genes, the presence of only few differentially expressed genes and/or few outliers. Moreover, the cut-off for selecting significantly differentially expressed genes for further downstream analysis often depend on arbitrary choices.
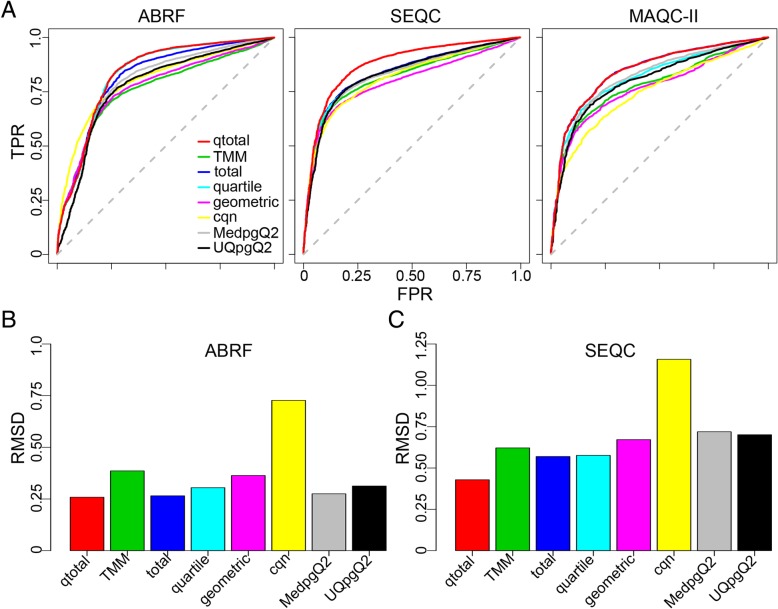
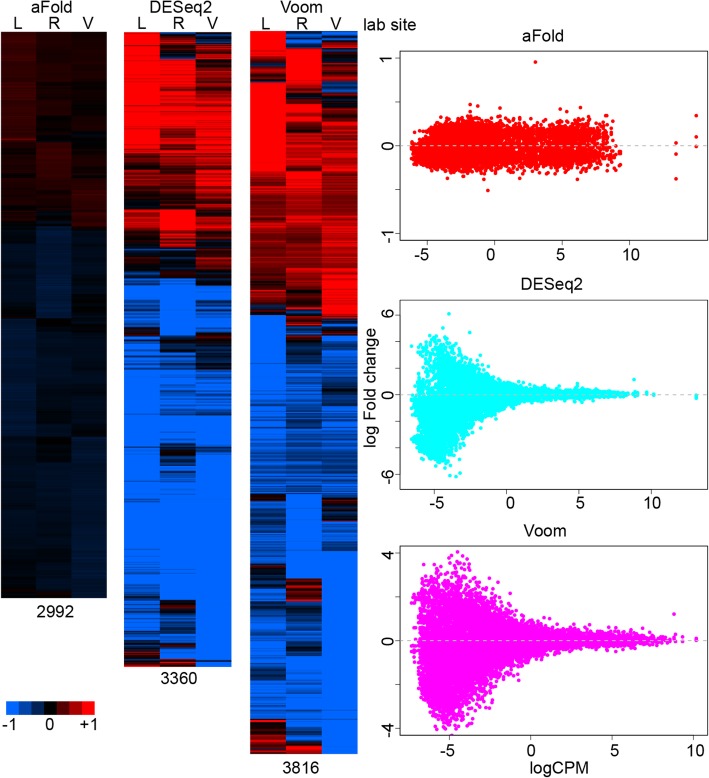
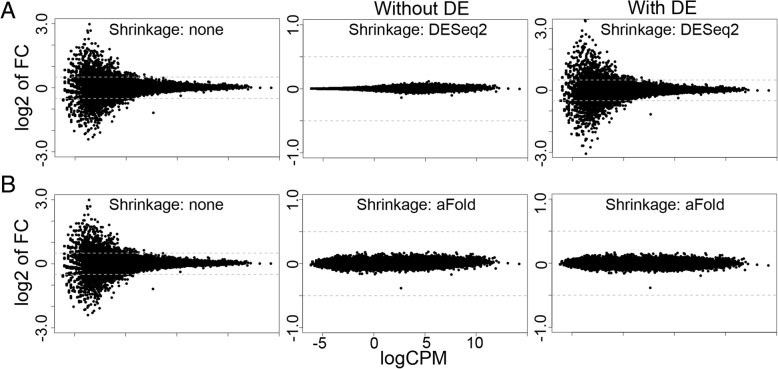
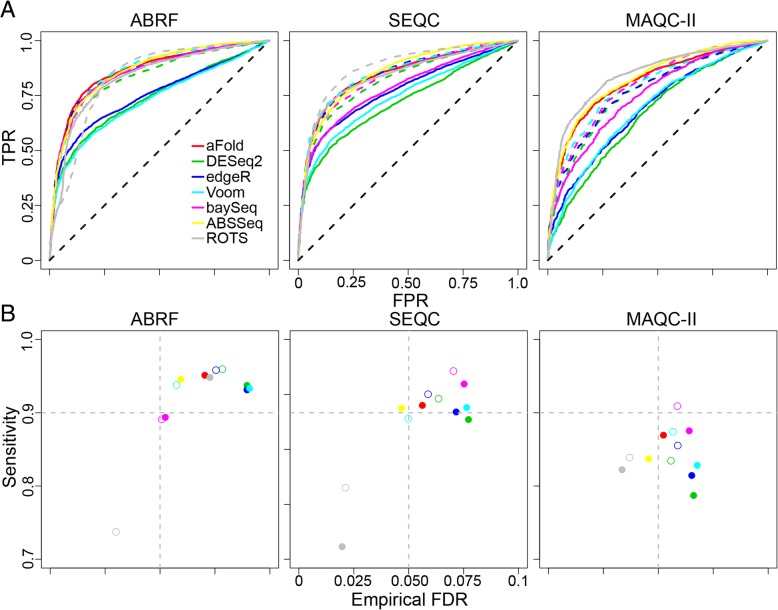
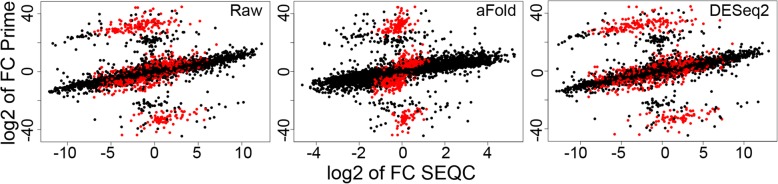
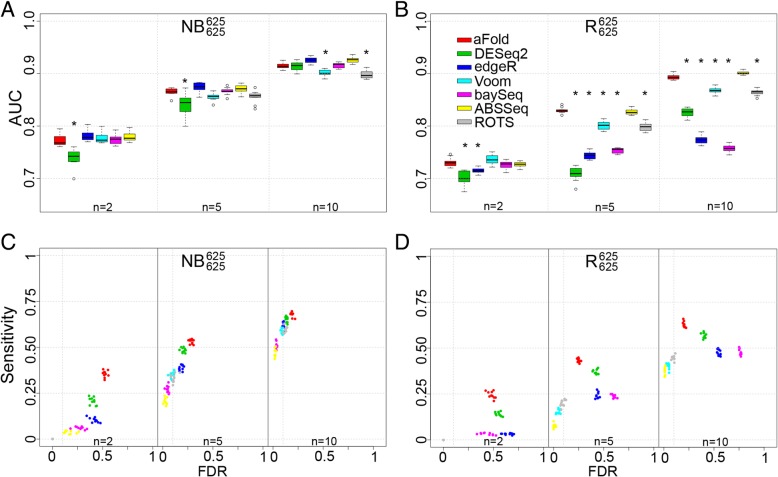
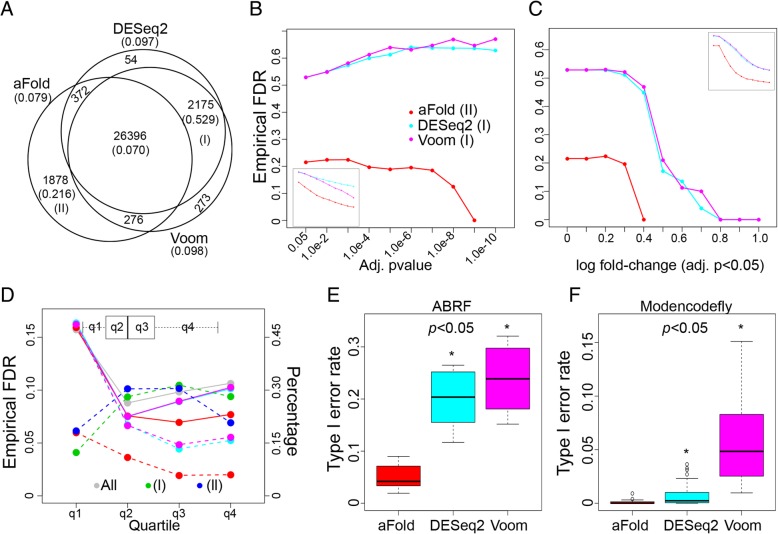
We here introduce a new tool for estimating differential expression in noisy real-life data. It employs a novel normalization procedure (qtotal), which takes account of the overall distribution of read counts for data standardization enhancing reliable identification of differential gene expression, especially in case of asymmetrical distributions of up- and downregulated genes. The tool then introduces a polynomial algorithm (aFold) to model the uncertainty of read counts across treatments and genes. We extensively benchmark aFold on a variety of simulated and validated real-life data sets (e.g. ABRF, SEQC and MAQC-II) and show a higher ability to correctly identify differentially expressed genes under most tested conditions. aFold infers fold change values that are comparable across experiments, thereby facilitating data clustering, visualization, and other downstream applications.
We here present a new transcriptomics analysis tool that includes both a data normalization method and a differential expression analysis approach. The new tool is shown to enhance reliable identification of significant differential expression across distinct data distributions. It outcompetes alternative procedures in case of asymmetrical distributions of up- versus down-regulated genes and also the presence of outliers, all common to real data sets.
数据标准化和显著差异表达的鉴定是 RNA-Seq 分析的关键步骤。许多现有的工具都依赖于一些假设,而这些假设往往不符合实际数据,包括上调和下调基因分布对称、只有少数差异表达基因和/或少数离群值的常见假设。此外,选择用于进一步下游分析的显著差异表达基因的截止值通常取决于任意选择。
我们在这里介绍了一种用于估计嘈杂实际数据中差异表达的新工具。它采用了一种新的标准化程序(qtotal),考虑了读频数的总体分布,以增强差异基因表达的可靠鉴定,特别是在上调和下调基因分布不对称的情况下。然后,该工具引入了一种多项式算法(aFold)来模拟处理和基因之间读频数的不确定性。我们在各种模拟和验证的实际数据集(例如 ABRF、SEQC 和 MAQC-II)上广泛地对 aFold 进行基准测试,并在大多数测试条件下显示出更高的正确鉴定差异表达基因的能力。aFold 推断的倍数变化值在实验之间具有可比性,从而促进了数据聚类、可视化和其他下游应用。
我们在这里提出了一种新的转录组学分析工具,它包括数据标准化方法和差异表达分析方法。新工具显示,在不同的数据分布中,可靠地鉴定显著差异表达的能力得到了增强。在存在不对称的上调和下调基因分布以及离群值的情况下,它优于替代程序,这些都是实际数据集的常见情况。