Section of Pharmacology, Department of Pharmacy & Drug Sciences, University of Bari-Aldo Moro, Bari I-70125, Italy.
Section of Pharmacology, Department of Pharmacy & Drug Sciences, University of Bari-Aldo Moro, Bari I-70125, Italy.
Exp Neurol. 2014 May;255(100):96-102. doi: 10.1016/j.expneurol.2014.02.023. Epub 2014 Mar 5.
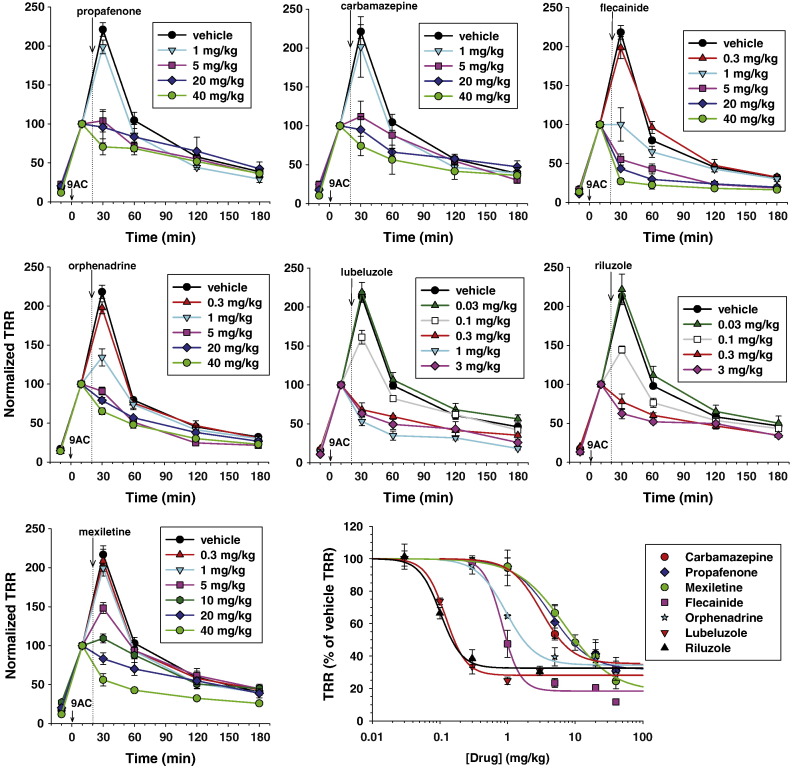
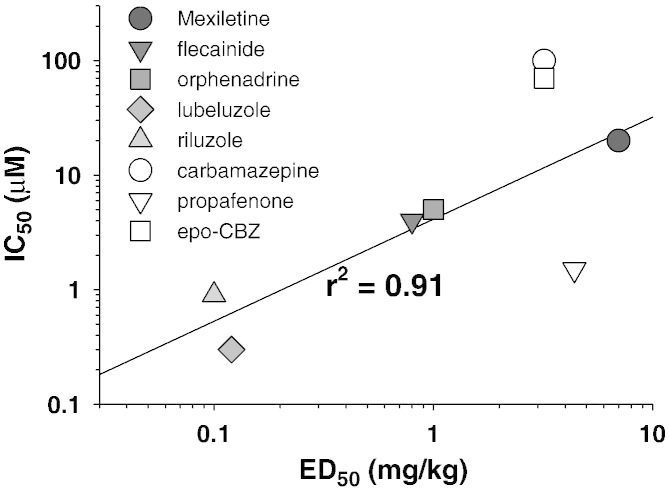
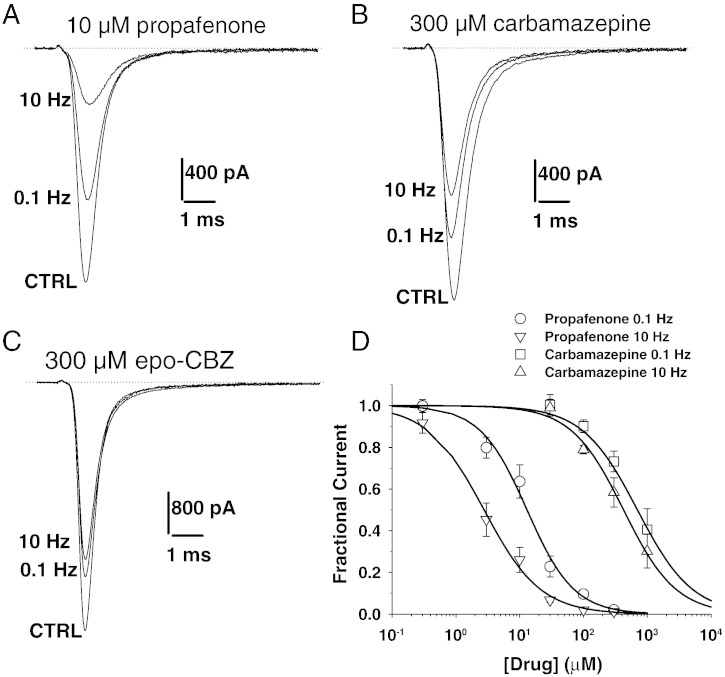
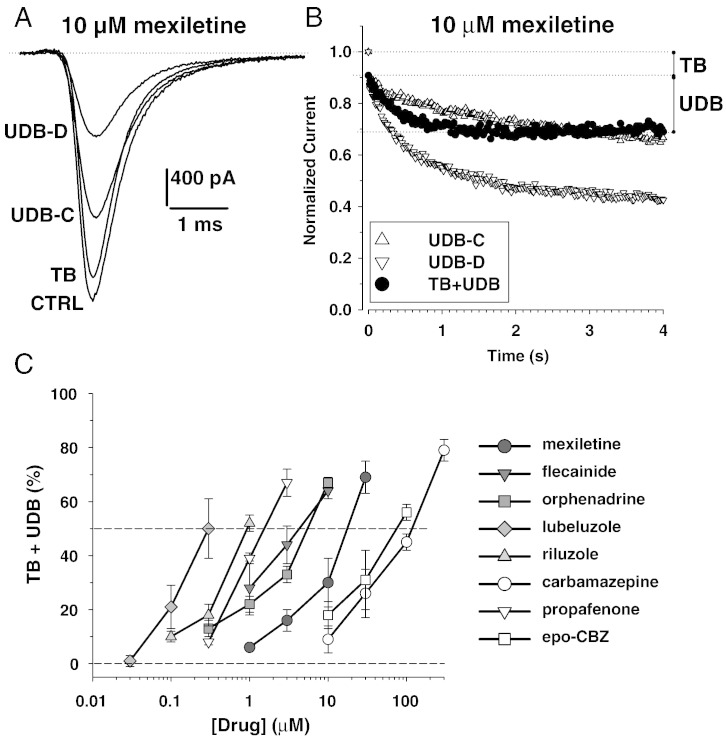
Although the sodium channel blocker mexiletine is considered the first-line drug in myotonia, some patients experiment adverse effects, while others do not gain any benefit. Other antimyotonic drugs are thus needed to offer mexiletine alternatives. In the present study, we used a previously-validated rat model of myotonia congenita to compare six marketed sodium channel blockers to mexiletine. Myotonia was induced in the rat by injection of anthracen-9-carboxylic acid, a muscle chloride channel blocker. The drugs were given orally and myotonia was evaluated by measuring the time of righting reflex. The drugs were also tested on sodium currents recorded in a cell line transfected with the human skeletal muscle sodium channel hNav1.4 using patch-clamp technique. In vivo, carbamazepine and propafenone showed antimyotonic activity at doses similar to mexiletine (ED50 close to 5mg/kg); flecainide and orphenadrine showed greater potency (ED50 near 1mg/kg); lubeluzole and riluzole were the more potent (ED50 near 0.1mg/kg). The antimyotonic activity of drugs in vivo was linearly correlated with their potency in blocking hNav1.4 channels in vitro. Deviation was observed for propafenone and carbamazepine, likely due to pharmacokinetics and multiple targets. The comparison of the antimyotonic dose calculated in rats with the current clinical dose in humans strongly suggests that all the tested drugs may be used safely for the treatment of human myotonia. Considering the limits of mexiletine tolerability and the occurrence of non-responders, this study proposes an arsenal of alternative drugs, which may prove useful to increase the quality of life of individuals suffering from non-dystrophic myotonia. Further clinical trials are warranted to confirm these results.
尽管钠离子通道阻滞剂美西律被认为是先天性肌强直的首选药物,但有些患者会出现不良反应,而有些患者则没有受益。因此,需要其他抗肌强直药物来替代美西律。在本研究中,我们使用了先前经过验证的先天性肌强直大鼠模型,将六种市售的钠离子通道阻滞剂与美西律进行了比较。通过注射蒽-9-羧酸(一种肌肉氯离子通道阻滞剂)在大鼠中诱导肌强直。将药物口服给予,并通过测量翻正反射的时间来评估肌强直。还使用膜片钳技术在转染人骨骼肌钠离子通道 hNav1.4 的细胞系上测试了这些药物对钠离子电流的作用。在体内,卡马西平和普罗帕酮在与美西律相似的剂量下表现出抗肌强直活性(ED50 接近 5mg/kg);氟卡尼和奥芬那君表现出更高的效力(ED50 接近 1mg/kg);卢贝鲁唑和利鲁唑的效力更强(ED50 接近 0.1mg/kg)。药物在体内的抗肌强直活性与其在体外阻断 hNav1.4 通道的效力呈线性相关。普罗帕酮和卡马西平的偏差可能是由于药代动力学和多种靶点所致。在大鼠中计算的抗肌强直剂量与当前临床剂量在人类中的比较强烈表明,所有测试的药物都可安全用于治疗人类肌强直。考虑到美西律耐受性的限制和无反应者的出现,这项研究提出了一系列替代药物,这可能有助于提高患有非营养不良性肌强直的个体的生活质量。需要进一步的临床试验来证实这些结果。