Zhou Xionghui, Liu Juan
School of computer, Wuhan University, Wuhan, China.
PLoS One. 2014 Mar 17;9(3):e92023. doi: 10.1371/journal.pone.0092023. eCollection 2014.
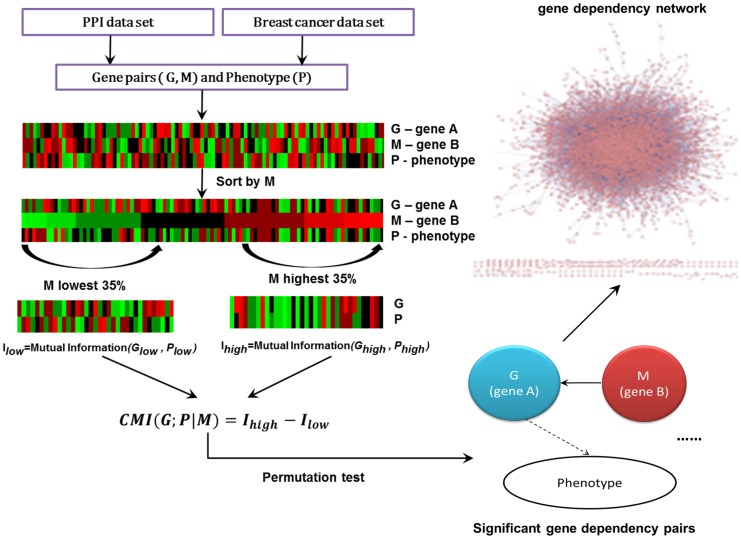
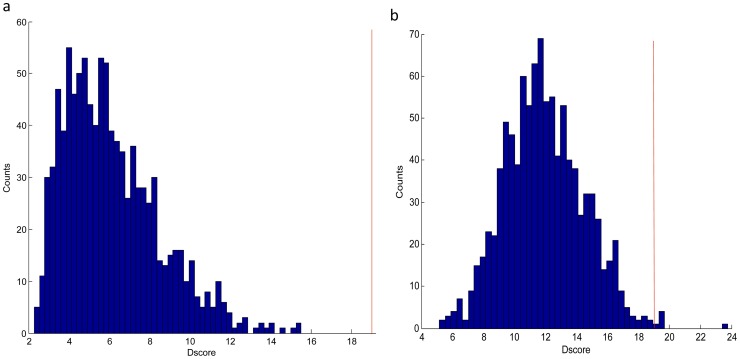
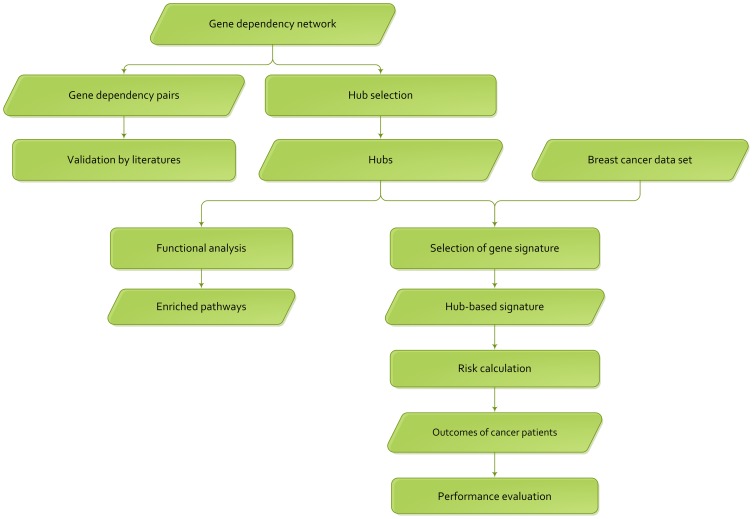
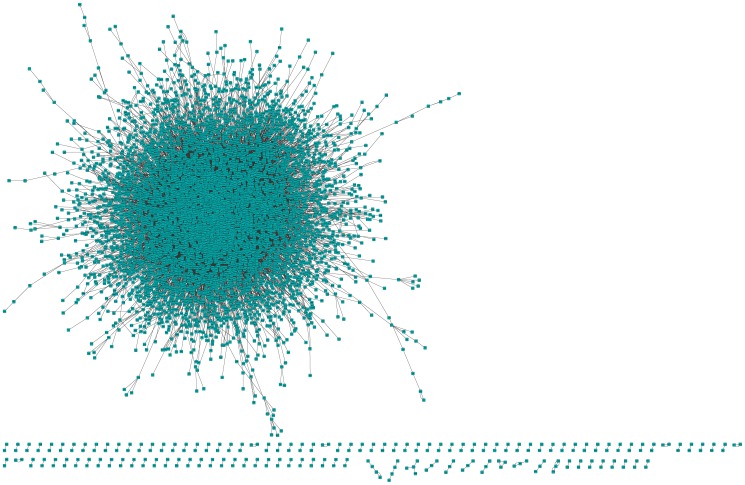
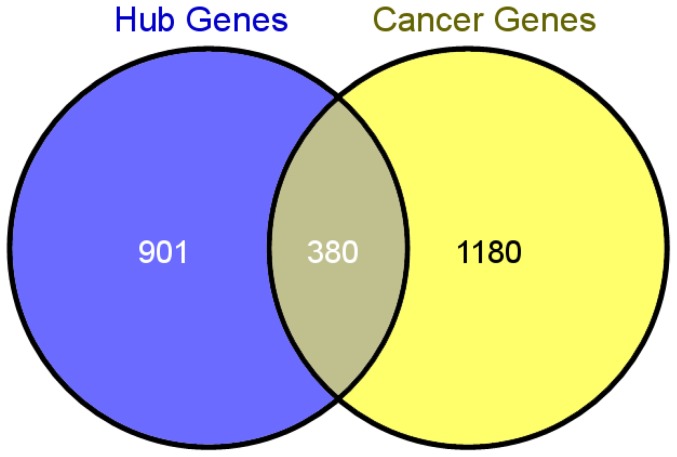
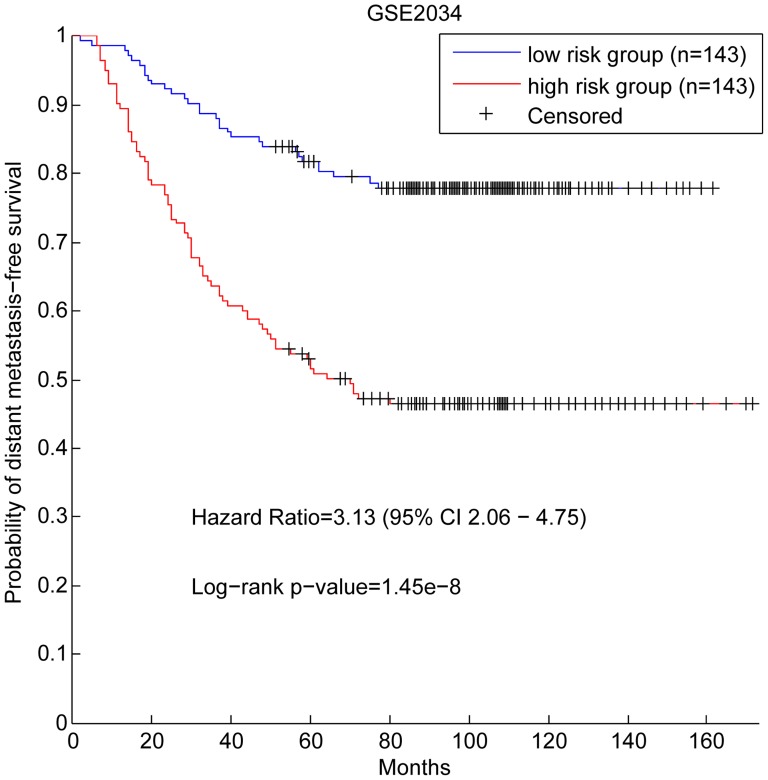
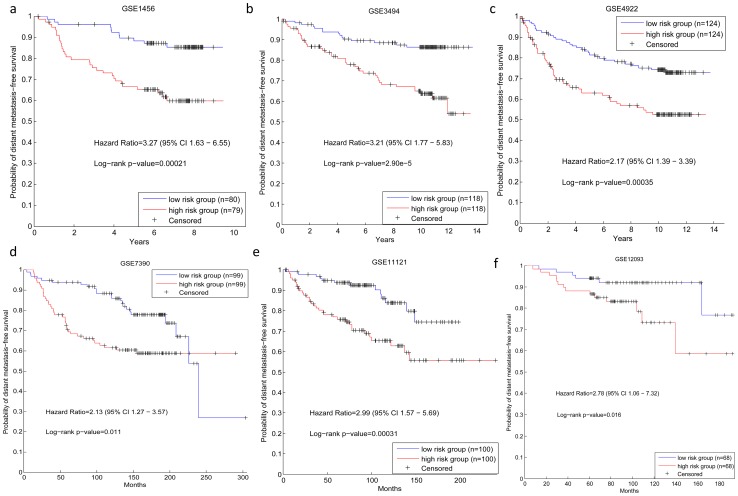
Although many methods have been proposed to reconstruct gene regulatory network, most of them, when applied in the sample-based data, can not reveal the gene regulatory relations underlying the phenotypic change (e.g. normal versus cancer). In this paper, we adopt phenotype as a variable when constructing the gene regulatory network, while former researches either neglected it or only used it to select the differentially expressed genes as the inputs to construct the gene regulatory network. To be specific, we integrate phenotype information with gene expression data to identify the gene dependency pairs by using the method of conditional mutual information. A gene dependency pair (A,B) means that the influence of gene A on the phenotype depends on gene B. All identified gene dependency pairs constitute a directed network underlying the phenotype, namely gene dependency network. By this way, we have constructed gene dependency network of breast cancer from gene expression data along with two different phenotype states (metastasis and non-metastasis). Moreover, we have found the network scale free, indicating that its hub genes with high out-degrees may play critical roles in the network. After functional investigation, these hub genes are found to be biologically significant and specially related to breast cancer, which suggests that our gene dependency network is meaningful. The validity has also been justified by literature investigation. From the network, we have selected 43 discriminative hubs as signature to build the classification model for distinguishing the distant metastasis risks of breast cancer patients, and the result outperforms those classification models with published signatures. In conclusion, we have proposed a promising way to construct the gene regulatory network by using sample-based data, which has been shown to be effective and accurate in uncovering the hidden mechanism of the biological process and identifying the gene signature for phenotypic change.
虽然已经提出了许多方法来重建基因调控网络,但其中大多数方法应用于基于样本的数据时,无法揭示表型变化(如正常与癌症)背后的基因调控关系。在本文中,我们在构建基因调控网络时将表型作为一个变量,而以往的研究要么忽略了它,要么仅将其用于选择差异表达基因作为构建基因调控网络的输入。具体而言,我们通过条件互信息方法将表型信息与基因表达数据相结合,以识别基因依赖对。基因依赖对(A,B)意味着基因A对表型的影响取决于基因B。所有识别出的基因依赖对构成了表型背后的一个有向网络,即基因依赖网络。通过这种方式,我们从基因表达数据以及两种不同的表型状态(转移和非转移)构建了乳腺癌的基因依赖网络。此外,我们发现该网络具有无标度性,这表明其出度高的枢纽基因可能在网络中发挥关键作用。经过功能研究,发现这些枢纽基因具有生物学意义且与乳腺癌特别相关,这表明我们的基因依赖网络是有意义的。文献研究也证明了其有效性。我们从该网络中选择了43个具有区分性的枢纽基因作为特征来构建区分乳腺癌患者远处转移风险的分类模型,其结果优于那些使用已发表特征的分类模型。总之,我们提出了一种利用基于样本的数据构建基因调控网络的有前景的方法,该方法在揭示生物过程的隐藏机制和识别表型变化的基因特征方面已被证明是有效且准确的。