Department of Computer Science and Engineering, National Taiwan Ocean University, Keelung, Taiwan.
BMC Bioinformatics. 2014 Apr 2;15:95. doi: 10.1186/1471-2105-15-95.
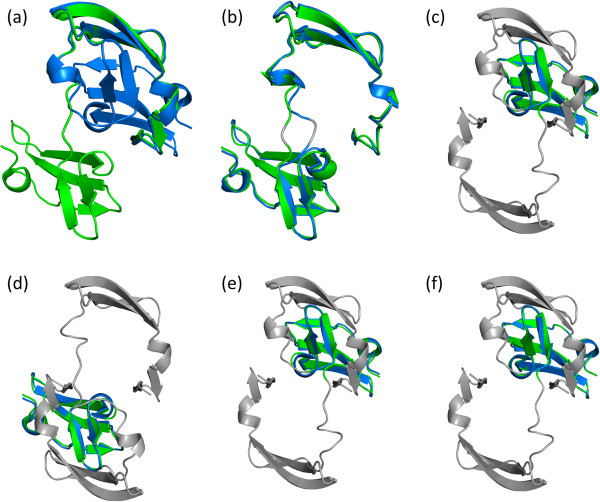
Protein structures are flexible and often show conformational changes upon binding to other molecules to exert biological functions. As protein structures correlate with characteristic functions, structure comparison allows classification and prediction of proteins of undefined functions. However, most comparison methods treat proteins as rigid bodies and cannot retrieve similarities of proteins with large conformational changes effectively.
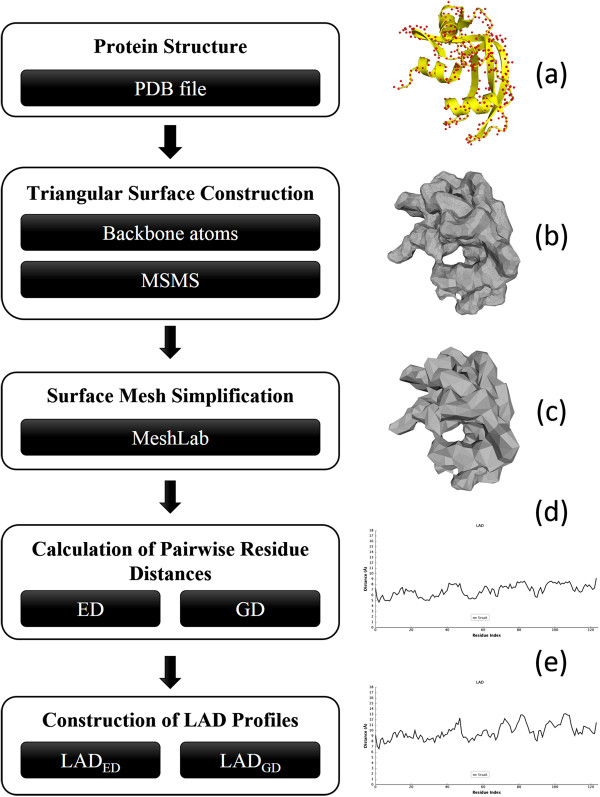
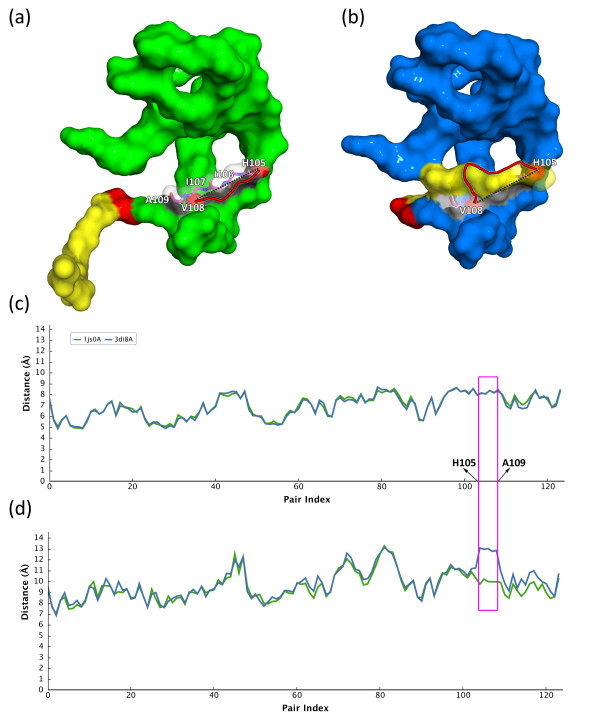
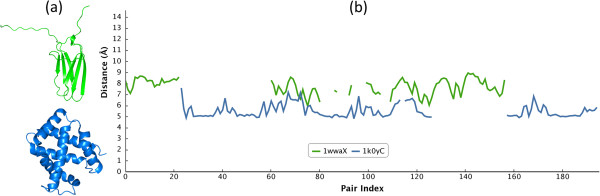
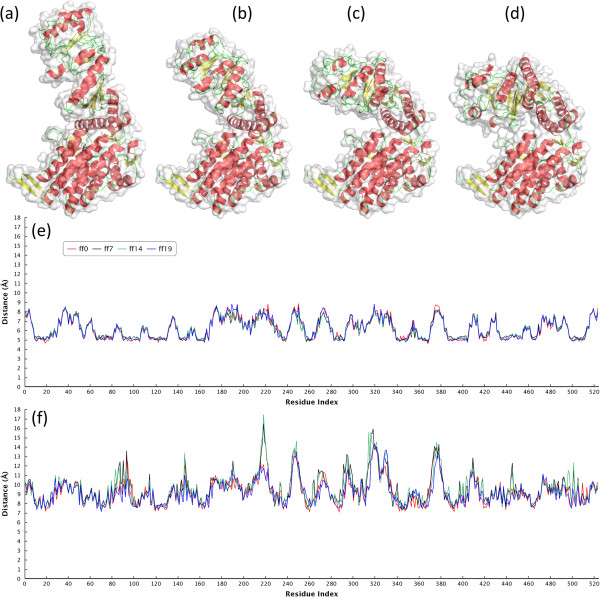
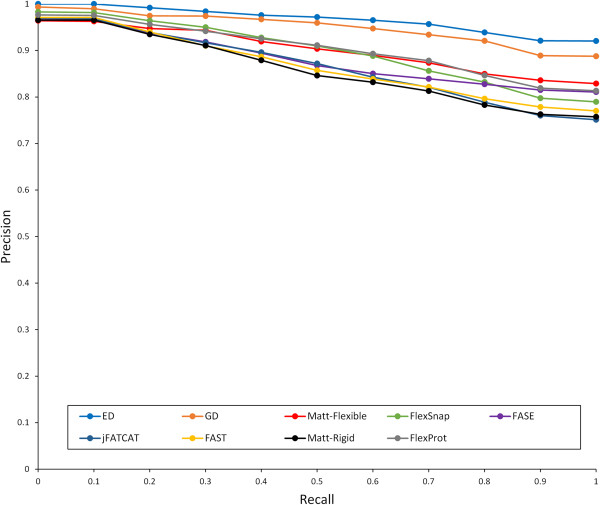
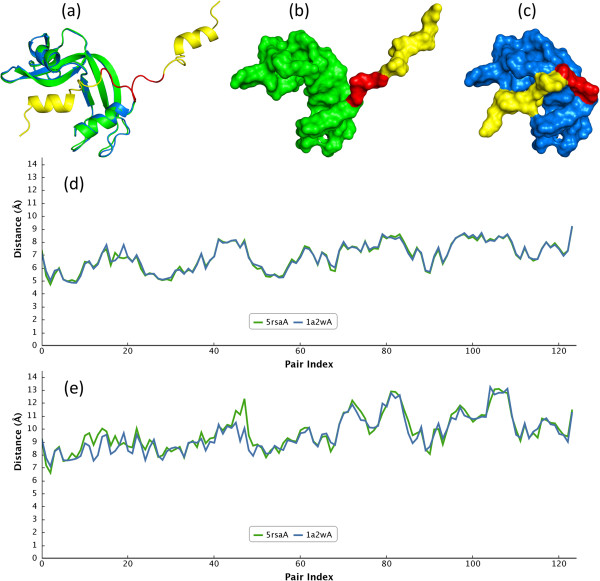
In this paper, we propose a novel descriptor, local average distance (LAD), based on either the geodesic distances (GDs) or Euclidean distances (EDs) for pairwise flexible protein structure comparison. The proposed method was compared with 7 structural alignment methods and 7 shape descriptors on two datasets comprising hinge bending motions from the MolMovDB, and the results have shown that our method outperformed all other methods regarding retrieving similar structures in terms of precision-recall curve, retrieval success rate, R-precision, mean average precision and F1-measure.
Both ED- and GD-based LAD descriptors are effective to search deformed structures and overcome the problems of self-connection caused by a large bending motion. We have also demonstrated that the ED-based LAD is more robust than the GD-based descriptor. The proposed algorithm provides an alternative approach for blasting structure database, discovering previously unknown conformational relationships, and reorganizing protein structure classification.
蛋白质结构具有柔韧性,在与其他分子结合时经常会发生构象变化,从而发挥生物功能。由于蛋白质结构与特征功能相关,因此结构比较可以对具有未知功能的蛋白质进行分类和预测。然而,大多数比较方法将蛋白质视为刚体,无法有效地检索具有较大构象变化的蛋白质的相似性。
在本文中,我们提出了一种新的描述符,基于测地距离(GDs)或欧几里得距离(EDs)的局部平均距离(LAD),用于成对的柔性蛋白质结构比较。该方法与 7 种结构比对方法和 7 种形状描述符在两个数据集上进行了比较,这两个数据集包含来自 MolMovDB 的铰链弯曲运动。结果表明,在精确召回曲线、检索成功率、R-精度、平均精度和 F1 度量方面,我们的方法在检索相似结构方面优于所有其他方法。
基于 ED 和 GD 的 LAD 描述符都可以有效地搜索变形结构,并克服由于大弯曲运动而导致的自连接问题。我们还证明了基于 ED 的 LAD 比基于 GD 的描述符更稳健。所提出的算法为爆破结构数据库、发现以前未知的构象关系以及重组蛋白质结构分类提供了一种替代方法。