INSERM, UMR-S 665, Dynamique des Structures et Interactions des Macromolécules Biologiques, Université Paris Diderot-Paris 7, Institut National de la Transfusion Sanguine, 6, rue Alexandre Cabanel, 75739 Paris cedex 15, France.
Nucleic Acids Res. 2011 Jul;39(Web Server issue):W18-23. doi: 10.1093/nar/gkr333. Epub 2011 May 17.
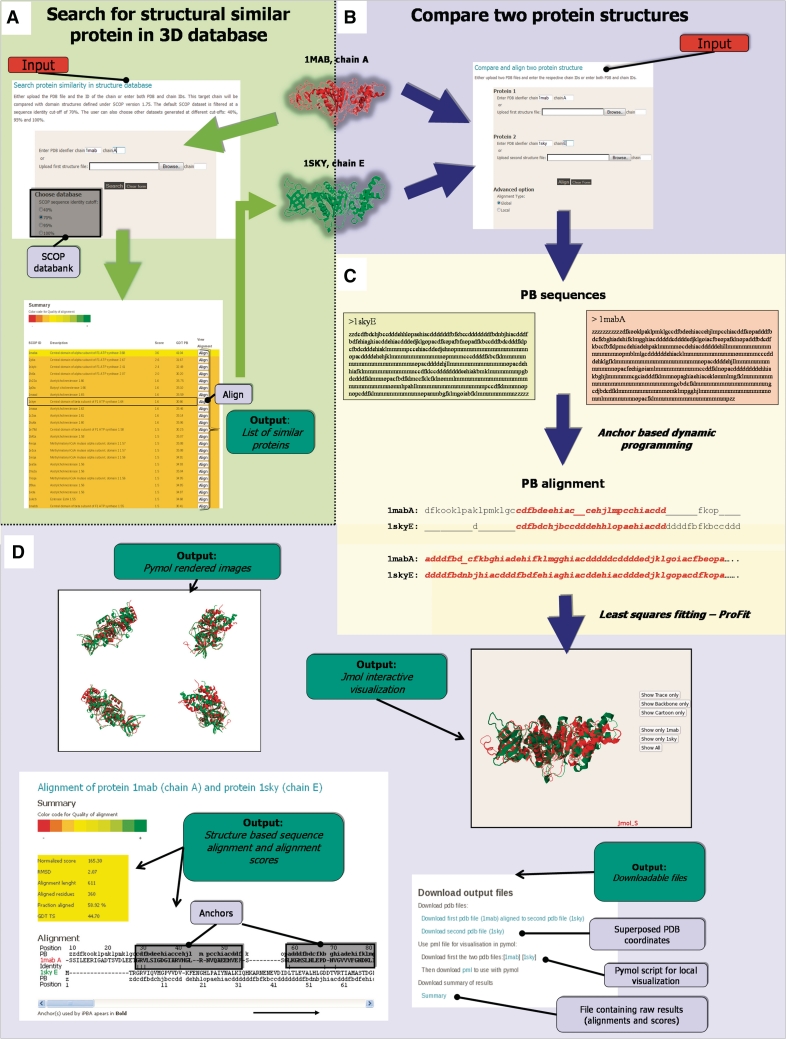
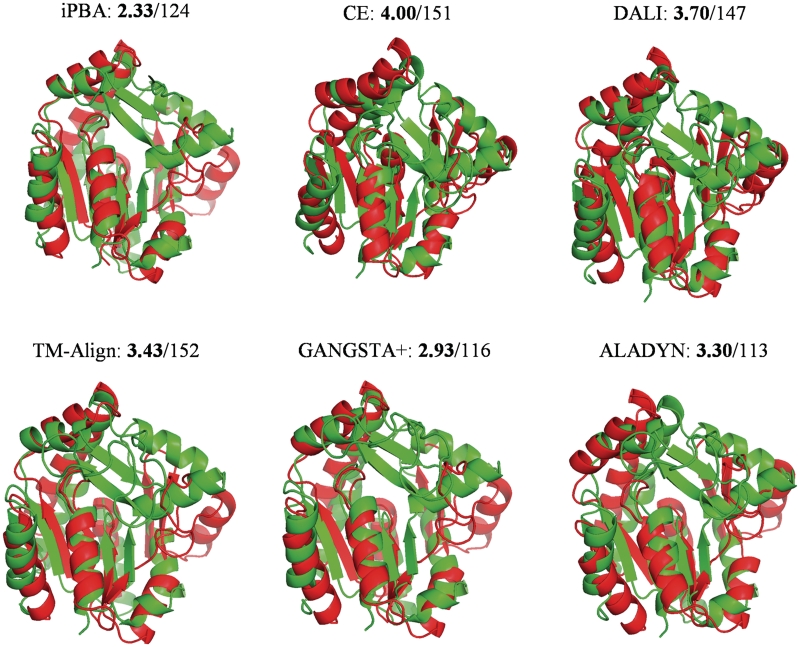
With the immense growth in the number of available protein structures, fast and accurate structure comparison has been essential. We propose an efficient method for structure comparison, based on a structural alphabet. Protein Blocks (PBs) is a widely used structural alphabet with 16 pentapeptide conformations that can fairly approximate a complete protein chain. Thus a 3D structure can be translated into a 1D sequence of PBs. With a simple Needleman-Wunsch approach and a raw PB substitution matrix, PB-based structural alignments were better than many popular methods. iPBA web server presents an improved alignment approach using (i) specialized PB Substitution Matrices (SM) and (ii) anchor-based alignment methodology. With these developments, the quality of ∼88% of alignments was improved. iPBA alignments were also better than DALI, MUSTANG and GANGSTA(+) in >80% of the cases. The webserver is designed to for both pairwise comparisons and database searches. Outputs are given as sequence alignment and superposed 3D structures displayed using PyMol and Jmol. A local alignment option for detecting subs-structural similarity is also embedded. As a fast and efficient 'sequence-based' structure comparison tool, we believe that it will be quite useful to the scientific community. iPBA can be accessed at http://www.dsimb.inserm.fr/dsimb_tools/ipba/.
随着可供使用的蛋白质结构数量的巨大增长,快速准确的结构比较已变得至关重要。我们提出了一种基于结构字母表的有效方法来进行结构比较。蛋白质块(PB)是一种广泛使用的结构字母表,它有 16 种五肽构象,可以相当近似地表示完整的蛋白质链。因此,三维结构可以转换为 PB 的一维序列。使用简单的 Needleman-Wunsch 方法和原始的 PB 替换矩阵,基于 PB 的结构比对优于许多流行的方法。iPBA 网络服务器使用(i)专门的 PB 替换矩阵(SM)和(ii)基于锚点的对齐方法呈现出一种改进的对齐方法。通过这些改进,约 88%的比对质量得到了提高。在超过 80%的情况下,iPBA 比对优于 DALI、MUSTANG 和 GANGSTA(+)。该网络服务器旨在进行两两比较和数据库搜索。输出以序列比对和使用 PyMol 和 Jmol 显示的叠加三维结构的形式给出。还嵌入了用于检测子结构相似性的局部比对选项。作为一种快速有效的“基于序列”的结构比较工具,我们相信它将对科学界非常有用。iPBA 可在 http://www.dsimb.inserm.fr/dsimb_tools/ipba/ 访问。