Laboratory of Molecular Neuroscience, Department of Biological Chemistry, The Edmond and Lily Safra Center of Brain Science, The Hebrew University of Jerusalem Jerusalem, Israel ; Bioinformatics, Genomnia srl Milano, Italy.
Laboratory of Molecular Neuroscience, Department of Biological Chemistry, The Edmond and Lily Safra Center of Brain Science, The Hebrew University of Jerusalem Jerusalem, Israel.
Front Cell Neurosci. 2014 Mar 27;8:89. doi: 10.3389/fncel.2014.00089. eCollection 2014.
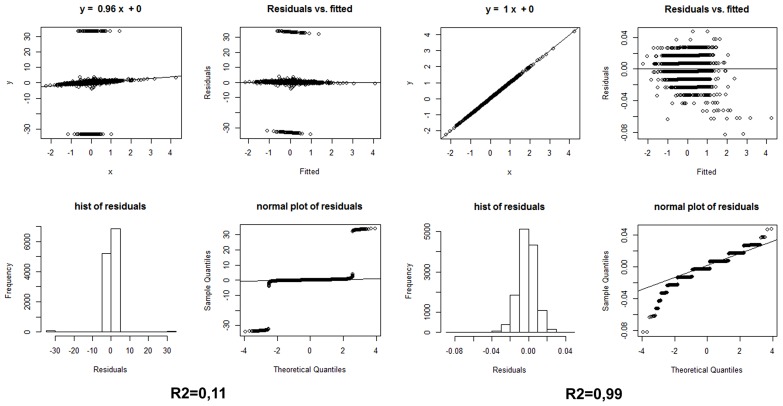
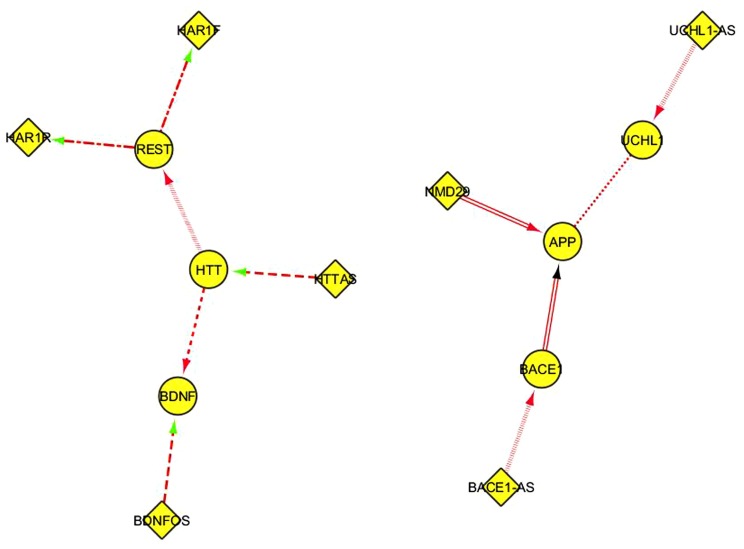
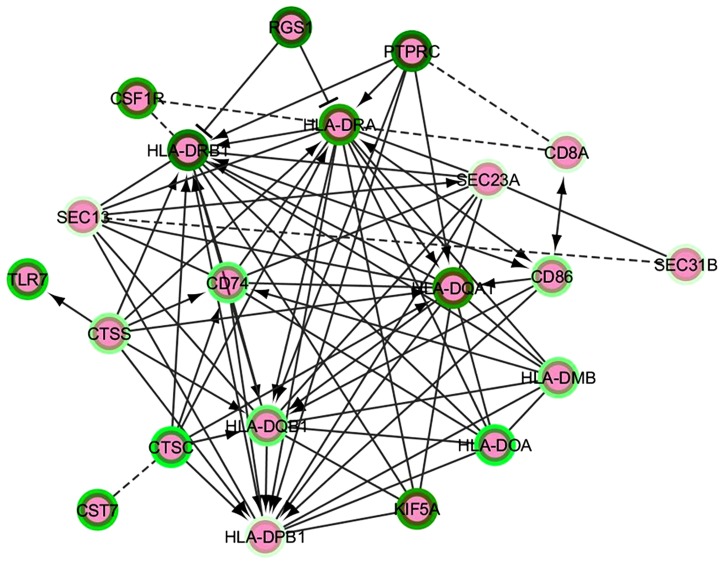
Neurodegenerative diseases in general and specifically late-onset Alzheimer's disease (LOAD) involve a genetically complex and largely obscure ensemble of causative and risk factors accompanied by complex feedback responses. The advent of "high-throughput" transcriptome investigation technologies such as microarray and deep sequencing is increasingly being combined with sophisticated statistical and bioinformatics analysis methods complemented by knowledge-based approaches such as Bayesian Networks or network and graph analyses. Together, such "integrative" studies are beginning to identify co-regulated gene networks linked with biological pathways and potentially modulating disease predisposition, outcome, and progression. Specifically, bioinformatics analyses of integrated microarray and genotyping data in cases and controls reveal changes in gene expression of both protein-coding and small and long regulatory RNAs; highlight relevant quantitative transcriptional differences between LOAD and non-demented control brains and demonstrate reconfiguration of functionally meaningful molecular interaction structures in LOAD. These may be measured as changes in connectivity in "hub nodes" of relevant gene networks (Zhang etal., 2013). We illustrate here the open analytical questions in the transcriptome investigation of neurodegenerative disease studies, proposing "ad hoc" strategies for the evaluation of differential gene expression and hints for a simple analysis of the non-coding RNA (ncRNA) part of such datasets. We then survey the emerging role of long ncRNAs (lncRNAs) in the healthy and diseased brain transcriptome and describe the main current methods for computational modeling of gene networks. We propose accessible modular and pathway-oriented methods and guidelines for bioinformatics investigations of whole transcriptome next generation sequencing datasets. We finally present methods and databases for functional interpretations of lncRNAs and propose a simple heuristic approach to visualize and represent physical and functional interactions of the coding and non-coding components of the transcriptome. Integrating in a functional and integrated vision coding and ncRNA analyses is of utmost importance for current and future analyses of neurodegenerative transcriptomes.
一般来说,神经退行性疾病,特别是迟发性阿尔茨海默病 (LOAD),涉及到一系列遗传上复杂且大部分未知的致病和风险因素,并伴有复杂的反馈反应。“高通量”转录组研究技术(如微阵列和深度测序)的出现,越来越多地与复杂的统计和生物信息学分析方法相结合,并辅以贝叶斯网络或网络和图形分析等基于知识的方法。这些“综合”研究开始一起识别与生物学途径相关的受共同调控的基因网络,并可能调节疾病易感性、结果和进展。具体来说,病例和对照的综合微阵列和基因分型数据分析的生物信息学分析揭示了蛋白质编码和小长调控 RNA 的基因表达变化;突出了 LOAD 和非痴呆对照大脑之间的相关定量转录差异,并证明了 LOAD 中功能有意义的分子相互作用结构的重新配置。这些可以作为相关基因网络“枢纽节点”的连接性变化来衡量(Zhang 等人,2013 年)。我们在这里说明了神经退行性疾病研究转录组研究中的开放性分析问题,提出了评估差异基因表达的“特定”策略,并为这些数据集的非编码 RNA (ncRNA) 部分提供了简单分析的提示。然后,我们调查了长 ncRNA (lncRNA) 在健康和患病大脑转录组中的新兴作用,并描述了基因网络计算建模的主要当前方法。我们提出了可访问的模块化和途径导向方法和指南,用于全转录组下一代测序数据集的生物信息学研究。最后,我们介绍了 lncRNA 功能解释的方法和数据库,并提出了一种简单的启发式方法来可视化和表示转录组的编码和非编码成分的物理和功能相互作用。在功能和综合视角下整合编码和 ncRNA 分析对于当前和未来的神经退行性转录组分析至关重要。