Giantin Mery, Granato Anna, Baratto Chiara, Marconato Laura, Vascellari Marta, Morello Emanuela M, Vercelli Antonella, Mutinelli Franco, Dacasto Mauro
Dipartimento di Biomedicina Comparata e Alimentazione, Università di Padova, Legnaro (Padova), Italy.
Istituto Zooprofilattico Sperimentale delle Venezie, Legnaro (Padova), Italy.
PLoS One. 2014 Apr 18;9(4):e95481. doi: 10.1371/journal.pone.0095481. eCollection 2014.
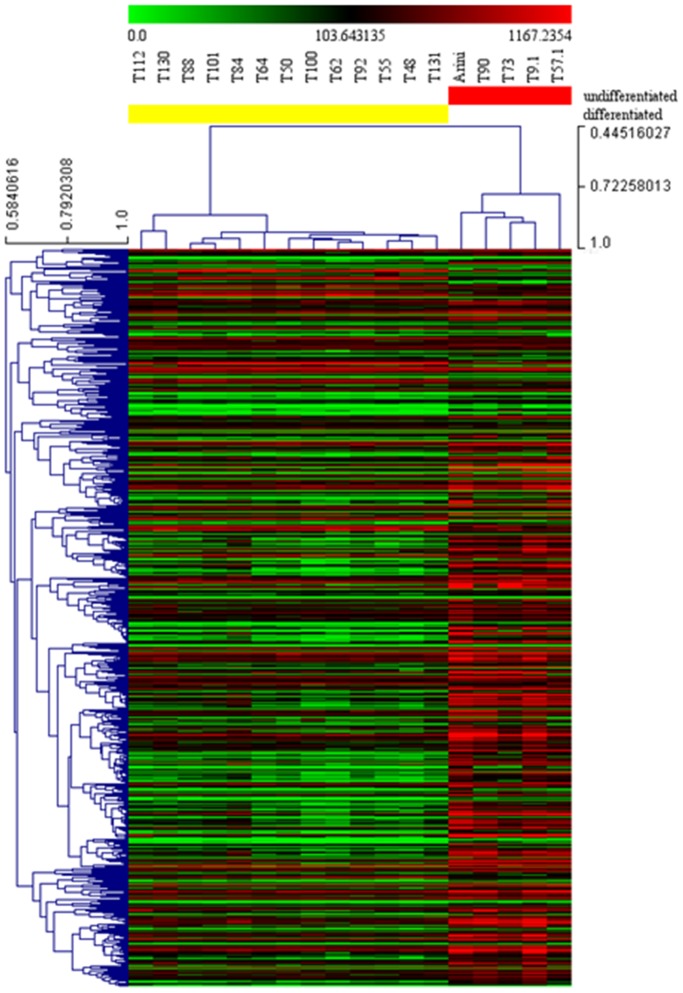
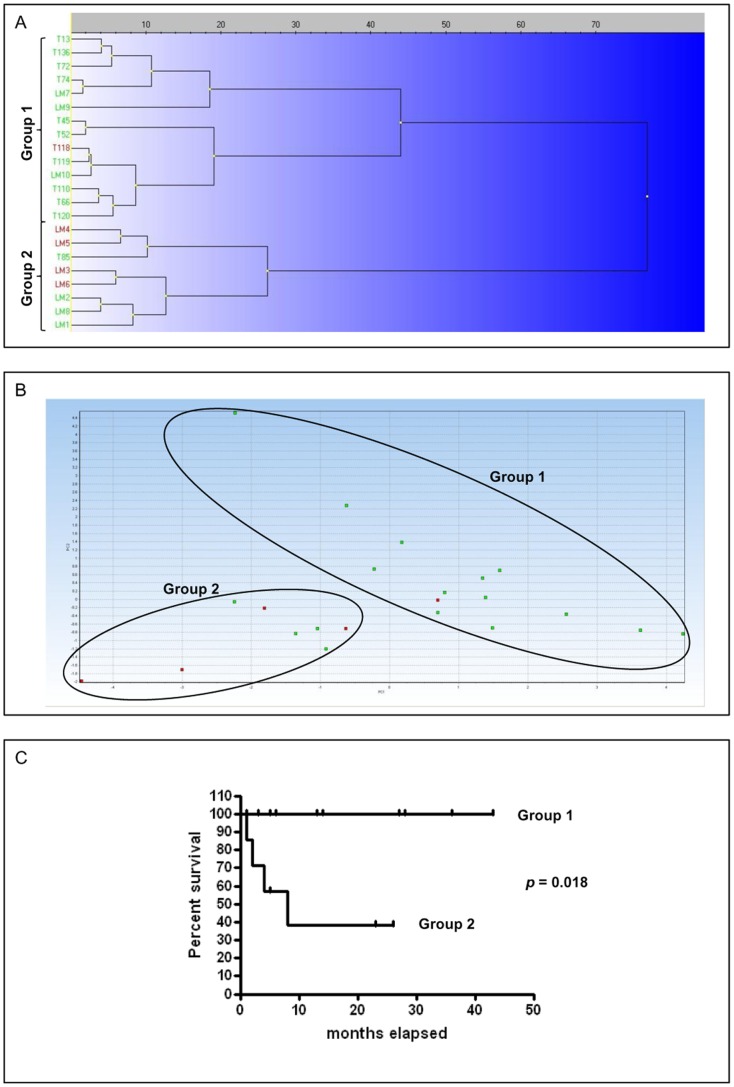
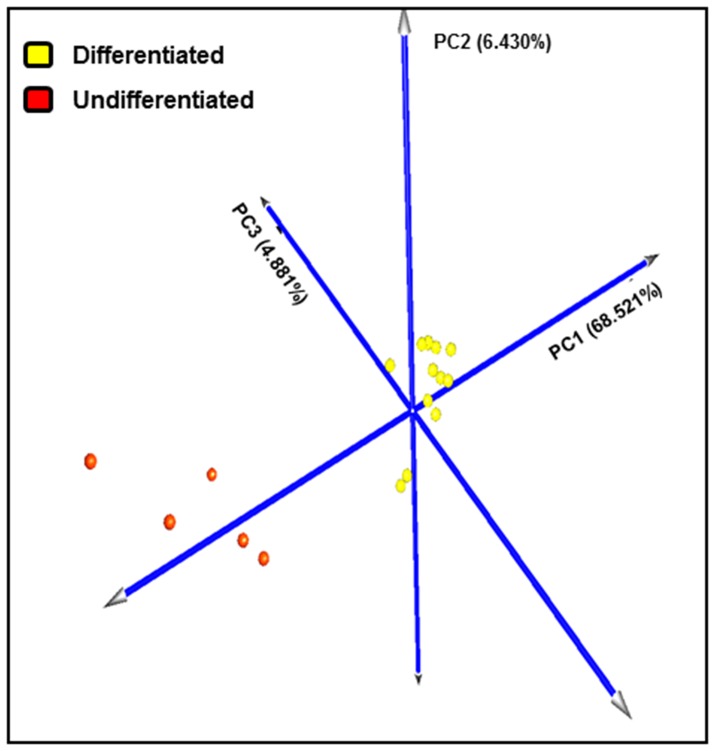
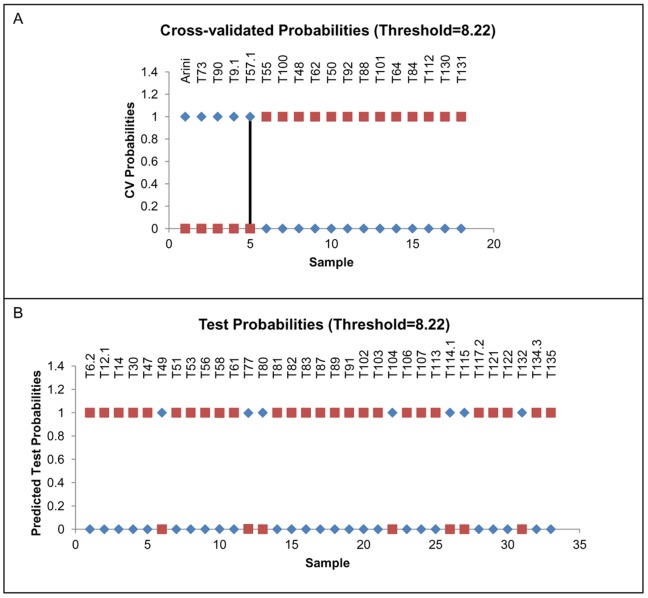
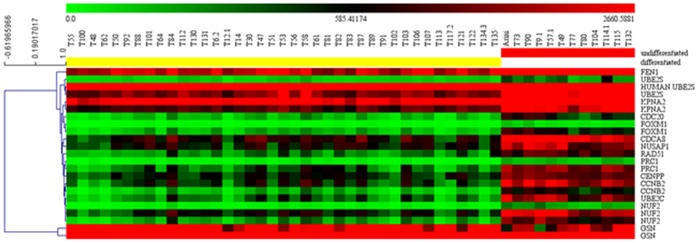
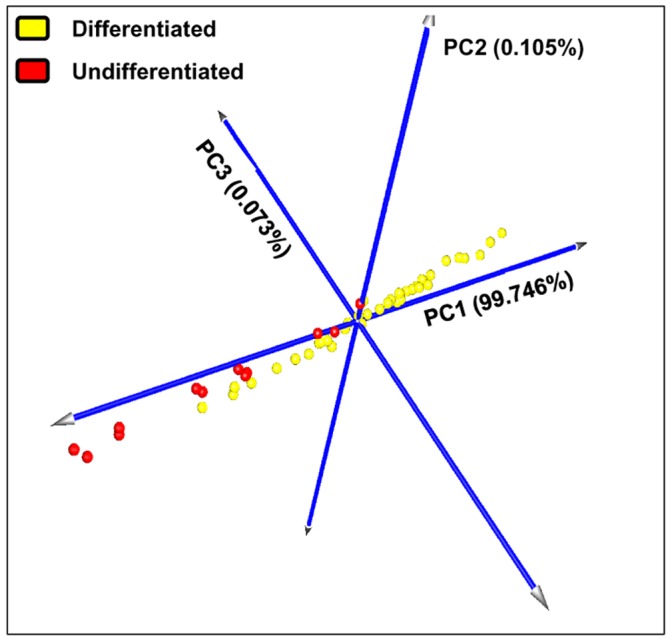
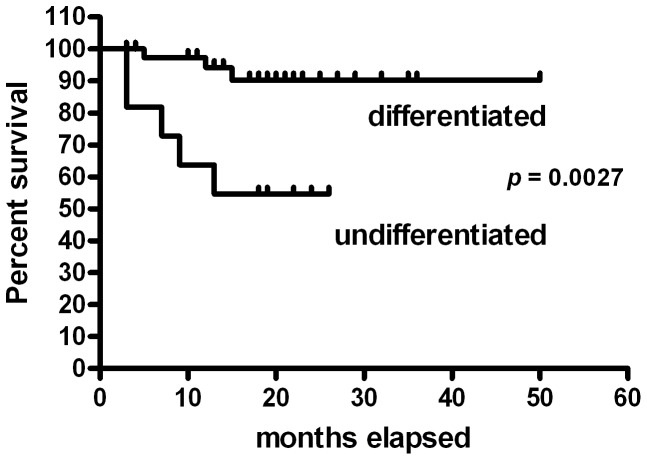
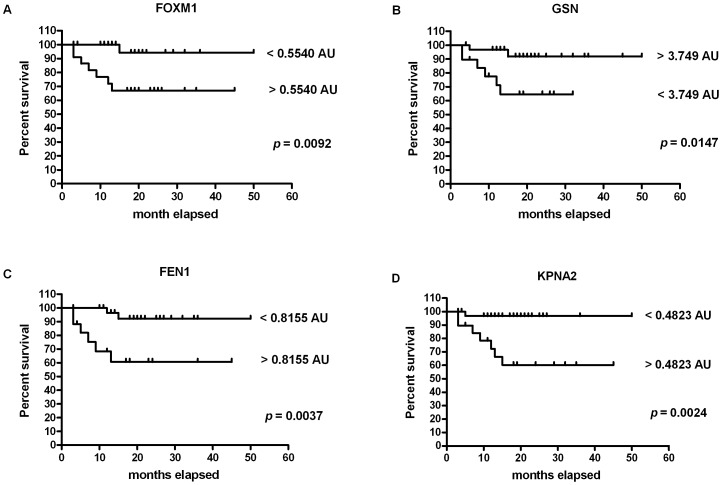
Prognosis and therapeutic management of dogs with cutaneous mast cell tumors (MCTs) depend on clinical stage and histological grade. However, the prognostic value of this latter is still questionable. In the present study, MCT transcriptome was analyzed to identify a set of candidate genes potentially useful for predicting the biological behavior of MCTs. Fifty-one canine MCT biopsies were analyzed. Isolated and purified total RNAs were individually hybridized to the Agilent Canine V2 4x44k DNA microarray. The comparison of reference differentiated and undifferentiated MCT transcriptome revealed a total of 597 differentially expressed genes (147 down-regulated and 450 up-regulated). The functional analysis of this set of genes provided evidence that they were mainly involved in cell cycle, DNA replication, p53 signaling pathway, nucleotide excision repair and pyrimidine metabolism. Class prediction analysis identified 13 transcripts providing the greatest accuracy of class prediction and divided samples into two categories (differentiated and undifferentiated), harboring a different prognosis. The Principal Component Analysis of all samples, made by using the selected 13 markers, confirmed MCT classification. The first three components accounted for 99.924% of the total variance. This molecular classification significantly correlated with survival time (p = 0.0026). Furthermore, among all marker genes, a significant association was found between mRNA expression and MCT-related mortality for FOXM1, GSN, FEN1 and KPNA2 (p<0.05). Finally, marker genes mRNA expression was evaluated in a cohort of 22 independent samples. Data obtained enabled to identify MCT cases with different prognosis. Overall, the molecular characterization of canine MCT transcriptome allowed the identification of a set of 13 transcripts that clearly separated differentiated from undifferentiated MCTs, thus predicting outcome regardless of the histological grade. These results may have clinical relevance and warrant future validation in a prospective study.
皮肤肥大细胞瘤(MCT)犬的预后及治疗管理取决于临床分期和组织学分级。然而,后者的预后价值仍存在疑问。在本研究中,对MCT转录组进行分析,以鉴定一组可能有助于预测MCT生物学行为的候选基因。分析了51份犬MCT活检样本。分离并纯化的总RNA分别与安捷伦犬V2 4x44k DNA微阵列杂交。参考分化型和未分化型MCT转录组的比较显示共有597个差异表达基因(147个下调和450个上调)。对这组基因的功能分析表明,它们主要参与细胞周期、DNA复制、p53信号通路、核苷酸切除修复和嘧啶代谢。类别预测分析确定了13个转录本,其提供了最高的类别预测准确性,并将样本分为两类(分化型和未分化型),具有不同的预后。使用选定的13个标志物对所有样本进行主成分分析,证实了MCT分类。前三个成分占总方差的99.924%。这种分子分类与生存时间显著相关(p = 0.0026)。此外,在所有标志物基因中,发现FOXM1、GSN、FEN1和KPNA2的mRNA表达与MCT相关死亡率之间存在显著关联(p<0.05)。最后,在一组22个独立样本中评估了标志物基因的mRNA表达。获得的数据能够识别具有不同预后的MCT病例。总体而言,犬MCT转录组的分子特征鉴定出一组13个转录本,可清晰区分分化型和未分化型MCT,从而无论组织学分级如何均可预测预后。这些结果可能具有临床相关性,值得在一项前瞻性研究中进行未来验证。