Tang Grace W, Altman Russ B
Department of Bioengineering, Stanford University, Stanford, California, United States of America.
Department of Bioengineering, Stanford University, Stanford, California, United States of America; Department of Genetics, Stanford University, Stanford, California, United States of America.
PLoS Comput Biol. 2014 Apr 24;10(4):e1003589. doi: 10.1371/journal.pcbi.1003589. eCollection 2014 Apr.
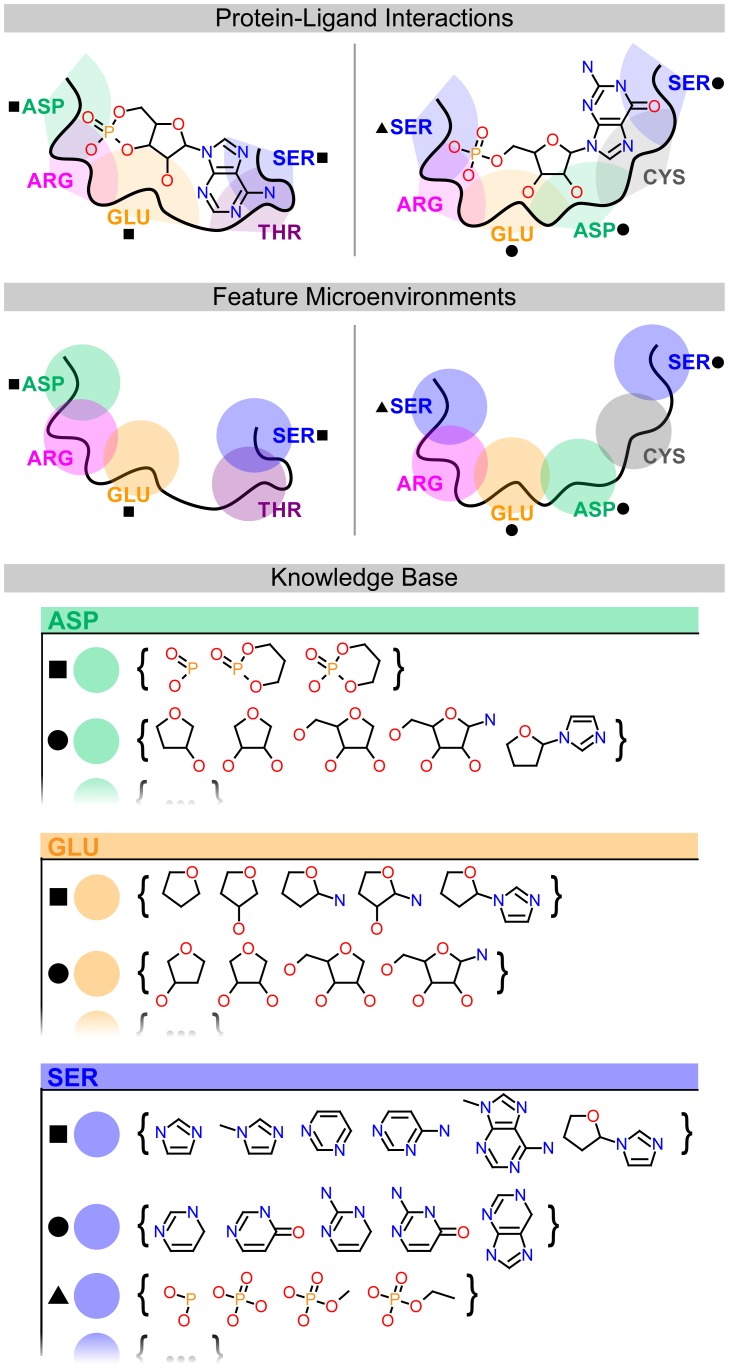
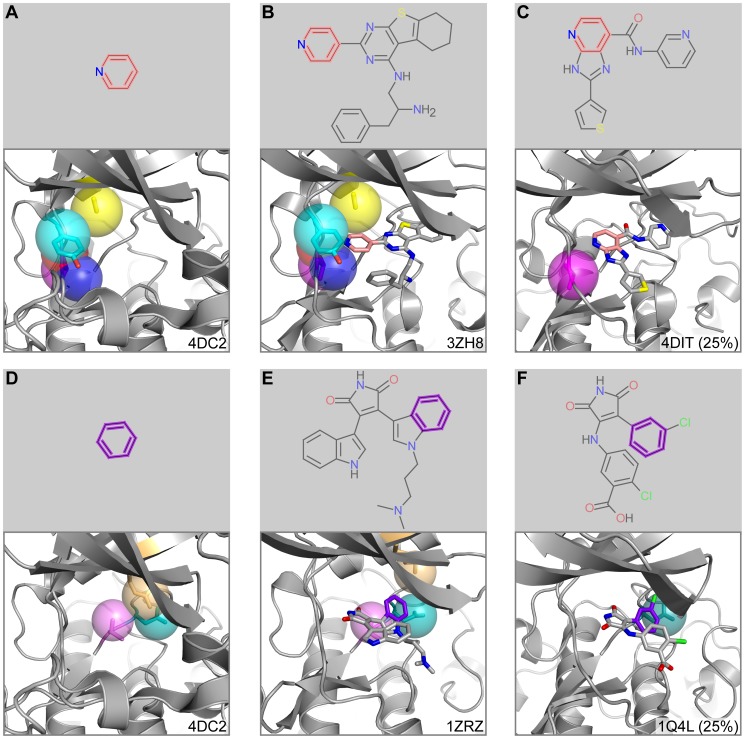
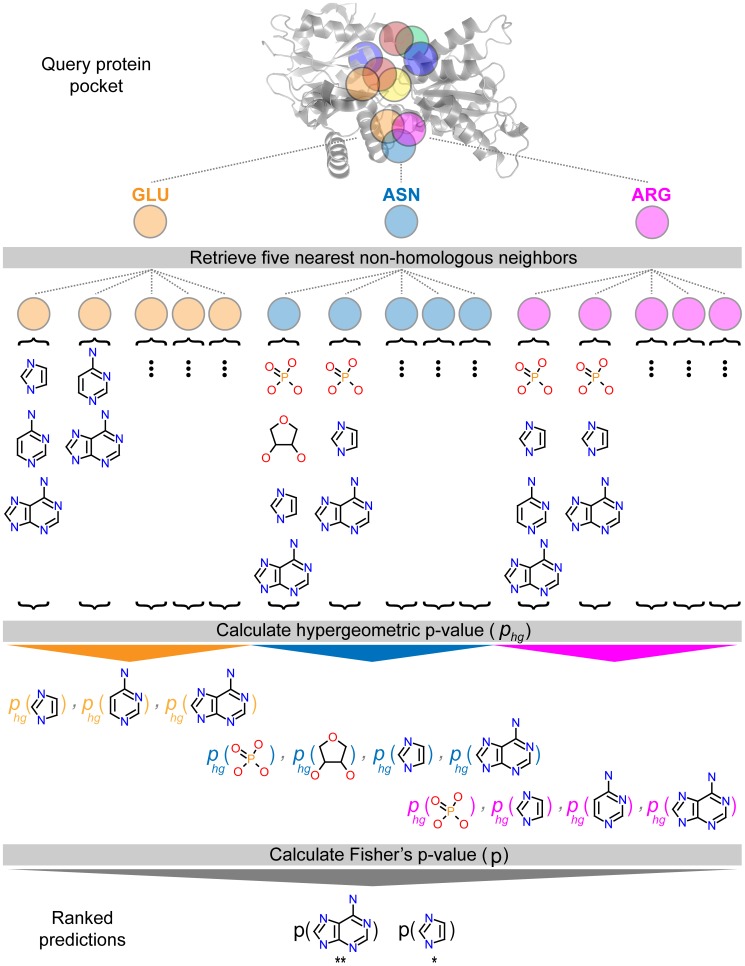
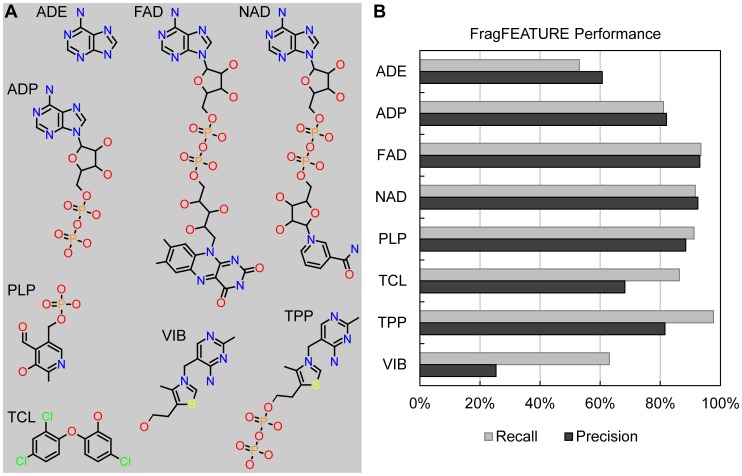
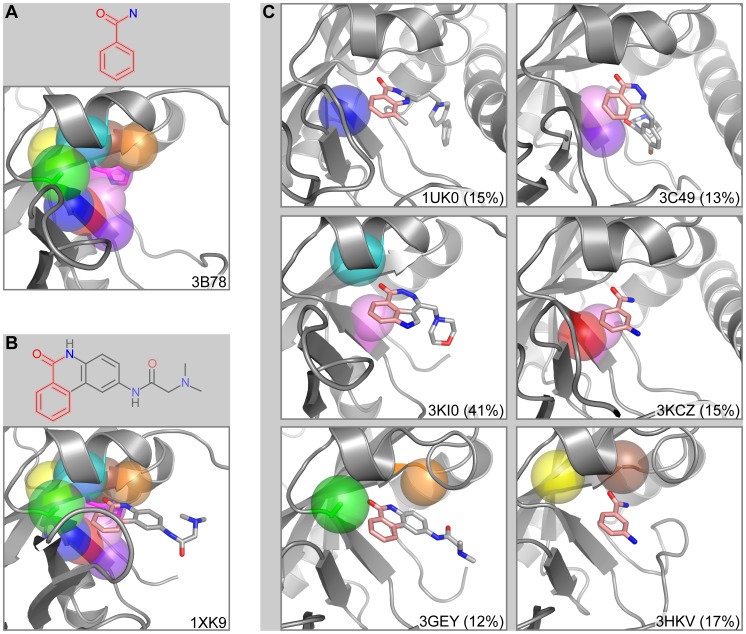
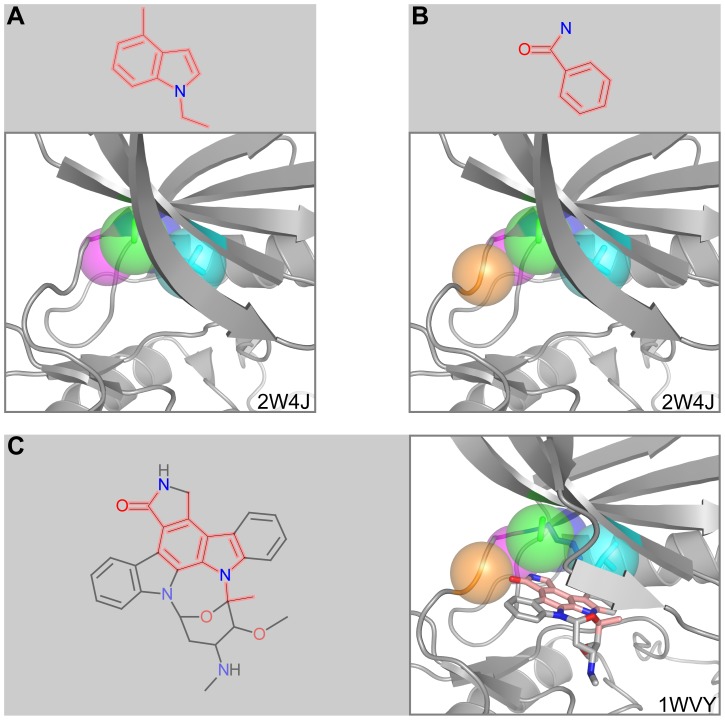
Target-based drug discovery must assess many drug-like compounds for potential activity. Focusing on low-molecular-weight compounds (fragments) can dramatically reduce the chemical search space. However, approaches for determining protein-fragment interactions have limitations. Experimental assays are time-consuming, expensive, and not always applicable. At the same time, computational approaches using physics-based methods have limited accuracy. With increasing high-resolution structural data for protein-ligand complexes, there is now an opportunity for data-driven approaches to fragment binding prediction. We present FragFEATURE, a machine learning approach to predict small molecule fragments preferred by a target protein structure. We first create a knowledge base of protein structural environments annotated with the small molecule substructures they bind. These substructures have low-molecular weight and serve as a proxy for fragments. FragFEATURE then compares the structural environments within a target protein to those in the knowledge base to retrieve statistically preferred fragments. It merges information across diverse ligands with shared substructures to generate predictions. Our results demonstrate FragFEATURE's ability to rediscover fragments corresponding to the ligand bound with 74% precision and 82% recall on average. For many protein targets, it identifies high scoring fragments that are substructures of known inhibitors. FragFEATURE thus predicts fragments that can serve as inputs to fragment-based drug design or serve as refinement criteria for creating target-specific compound libraries for experimental or computational screening.
基于靶点的药物发现必须评估许多具有潜在活性的类药物化合物。聚焦于低分子量化合物(片段)可以显著减少化学搜索空间。然而,确定蛋白质-片段相互作用的方法存在局限性。实验分析耗时、昂贵,且并非总是适用。同时,使用基于物理方法的计算方法准确性有限。随着蛋白质-配体复合物高分辨率结构数据的增加,现在有机会采用数据驱动的方法进行片段结合预测。我们提出了FragFEATURE,一种机器学习方法,用于预测目标蛋白质结构偏好的小分子片段。我们首先创建一个蛋白质结构环境知识库,用它们结合的小分子亚结构进行注释。这些亚结构分子量低,可作为片段的替代物。然后,FragFEATURE将目标蛋白质内的结构环境与知识库中的结构环境进行比较,以检索统计学上偏好的片段。它合并具有共享亚结构的不同配体的信息以生成预测。我们的结果表明,FragFEATURE能够重新发现与结合配体相对应的片段,平均精度为74%,召回率为82%。对于许多蛋白质靶点,它能识别出作为已知抑制剂亚结构的高分片段。因此,FragFEATURE预测的片段可作为基于片段的药物设计的输入,或作为创建用于实验或计算筛选的靶点特异性化合物库的优化标准。