Szöllősi Dániel, Horváth Tamás, Han Kyou-Hoon, Dokholyan Nikolay V, Tompa Péter, Kalmár Lajos, Hegedűs Tamás
MTA-SE Molecular Biophysics Research Group, Hungarian Academy of Sciences, Budapest, Hungary; Department of Biophysics and Radiation Biology, Semmelweis University, Budapest, Hungary.
Institute of Enzymology, Research Centre for Natural Sciences, Hungarian Academy of Sciences, Budapest, Hungary.
PLoS One. 2014 Apr 24;9(4):e95795. doi: 10.1371/journal.pone.0095795. eCollection 2014.
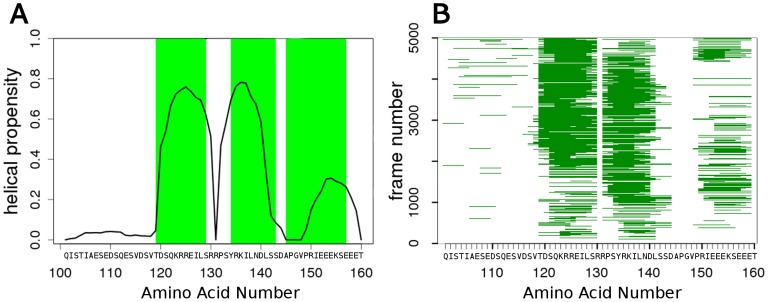
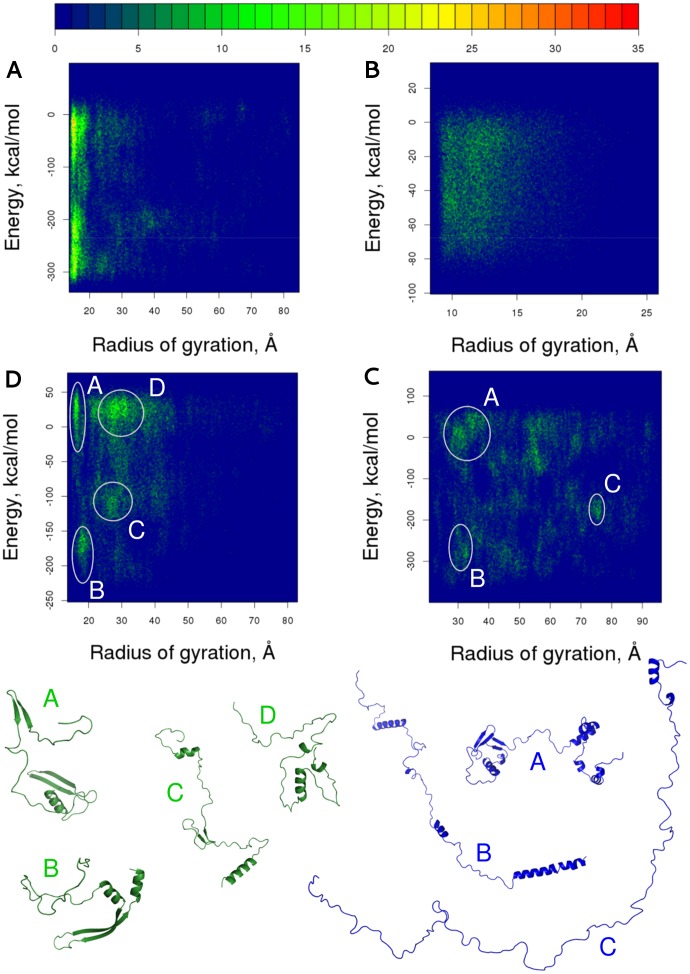
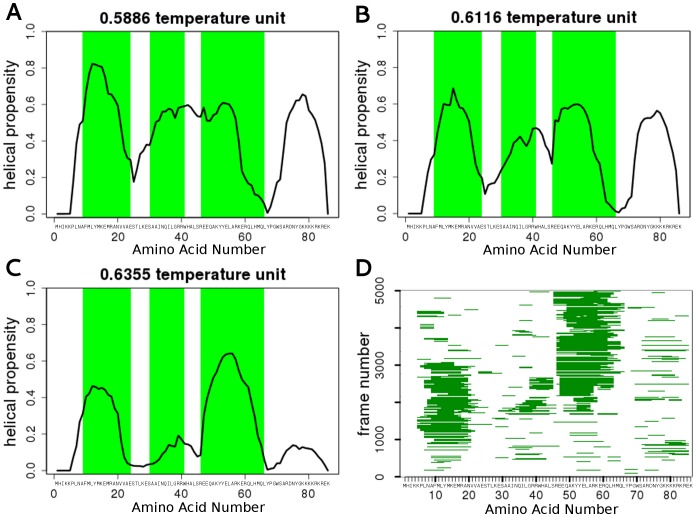
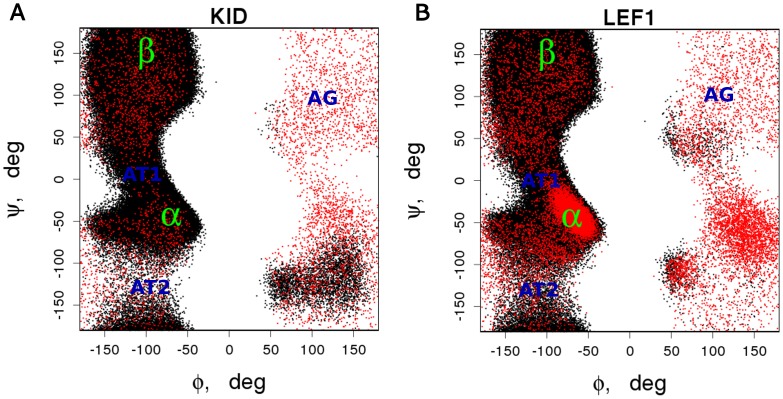
Intrinsically disordered proteins (IDPs) lack a stable tertiary structure, but their short binding regions termed Pre-Structured Motifs (PreSMo) can form transient secondary structure elements in solution. Although disordered proteins are crucial in many biological processes and designing strategies to modulate their function is highly important, both experimental and computational tools to describe their conformational ensembles and the initial steps of folding are sparse. Here we report that discrete molecular dynamics (DMD) simulations combined with replica exchange (RX) method efficiently samples the conformational space and detects regions populating α-helical conformational states in disordered protein regions. While the available computational methods predict secondary structural propensities in IDPs based on the observation of protein-protein interactions, our ab initio method rests on physical principles of protein folding and dynamics. We show that RX-DMD predicts α-PreSMos with high confidence confirmed by comparison to experimental NMR data. Moreover, the method also can dissect α-PreSMos in close vicinity to each other and indicate helix stability. Importantly, simulations with disordered regions forming helices in X-ray structures of complexes indicate that a preformed helix is frequently the binding element itself, while in other cases it may have a role in initiating the binding process. Our results indicate that RX-DMD provides a breakthrough in the structural and dynamical characterization of disordered proteins by generating the structural ensembles of IDPs even when experimental data are not available.
内在无序蛋白质(IDP)缺乏稳定的三级结构,但其被称为预结构化基序(PreSMo)的短结合区域可在溶液中形成瞬时二级结构元件。尽管无序蛋白质在许多生物过程中至关重要,并且设计调节其功能的策略非常重要,但用于描述其构象集合和折叠初始步骤的实验和计算工具都很匮乏。在此我们报告,离散分子动力学(DMD)模拟与副本交换(RX)方法相结合,能够有效地对构象空间进行采样,并检测无序蛋白质区域中形成α螺旋构象状态的区域。虽然现有的计算方法基于对蛋白质 - 蛋白质相互作用的观察来预测IDP中的二级结构倾向,但我们的从头算方法基于蛋白质折叠和动力学的物理原理。我们表明,通过与实验NMR数据比较,RX - DMD能够高度可靠地预测α - PreSMo。此外,该方法还可以剖析彼此紧邻的α - PreSMo,并表明螺旋稳定性。重要的是,对复合物X射线结构中形成螺旋的无序区域进行的模拟表明,预先形成的螺旋通常本身就是结合元件,而在其他情况下它可能在启动结合过程中发挥作用。我们的结果表明,RX - DMD通过生成IDP的结构集合,在无序蛋白质的结构和动力学表征方面取得了突破,即使在没有实验数据的情况下也是如此。