School of Computer Science, University of Birmingham Birmingham, UK.
Department Physiology and Pharmacology, State University of New York Downstate Medical Center Brooklyn, NY, USA ; Department Neurobiology, Yale University School of Medicine New Haven, CT, USA.
Front Comput Neurosci. 2014 Apr 3;8:39. doi: 10.3389/fncom.2014.00039. eCollection 2014.
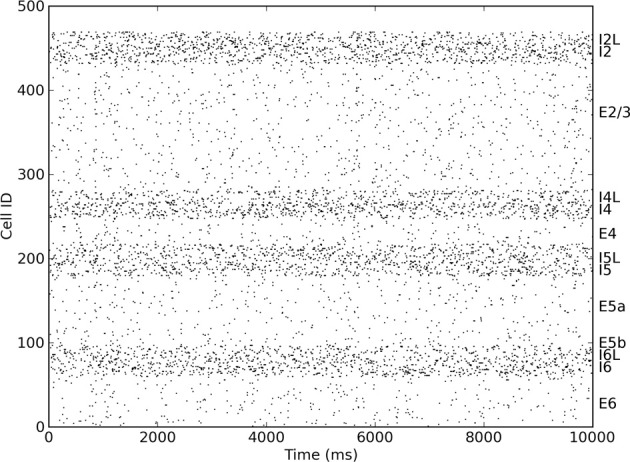
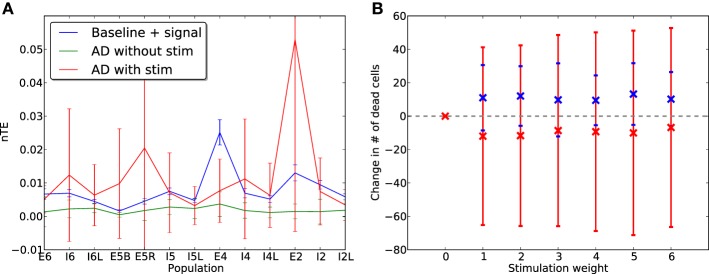
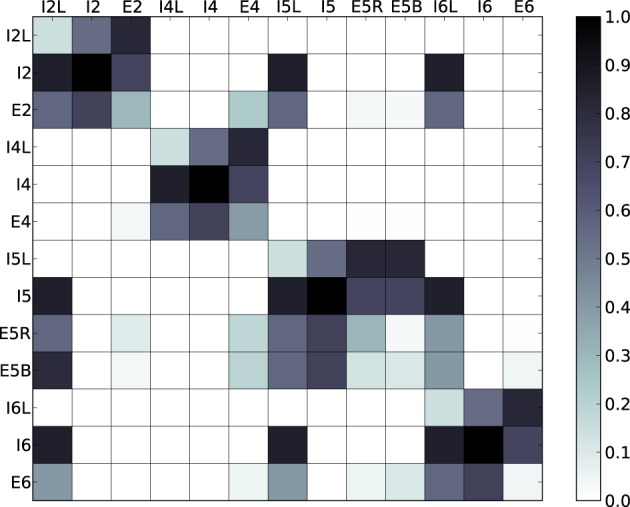
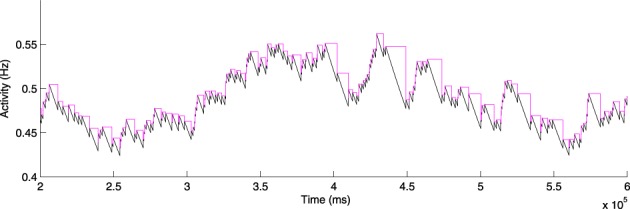
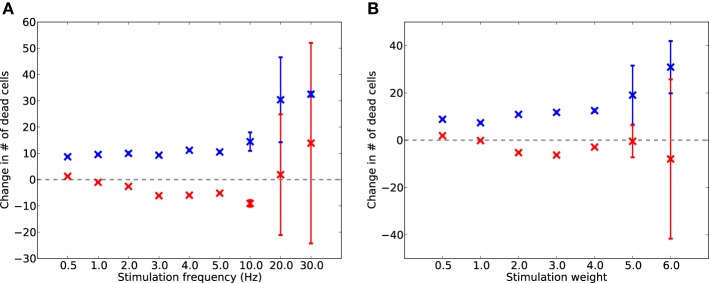
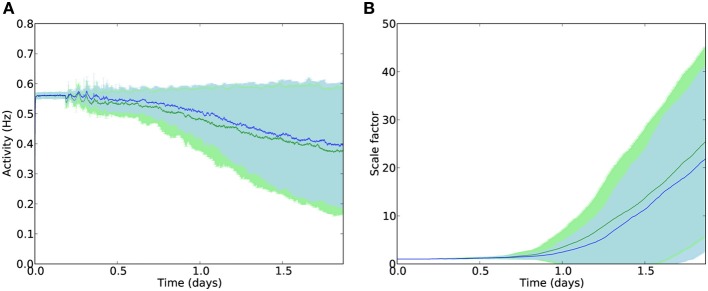
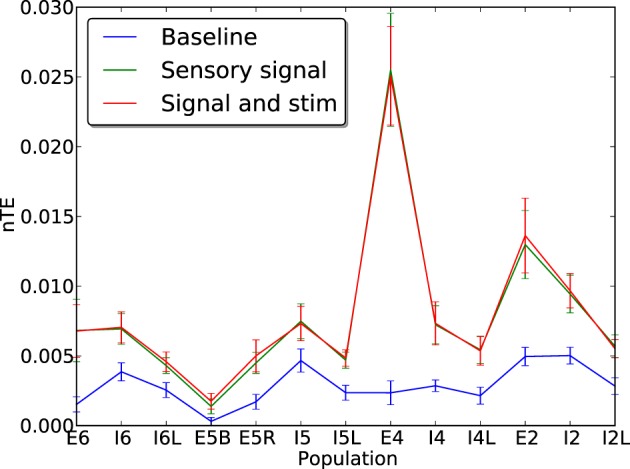
Cell death and synapse dysfunction are two likely causes of cognitive decline in AD. As cells die and synapses lose their drive, remaining cells suffer an initial decrease in activity. Neuronal homeostatic synaptic scaling then provides a feedback mechanism to restore activity. This homeostatic mechanism is believed to sense levels of activity-dependent cytosolic calcium within the cell and to adjust neuronal firing activity by increasing the density of AMPA synapses at remaining synapses to achieve balance. The scaling mechanism increases the firing rates of remaining cells in the network to compensate for decreases in network activity. However, this effect can itself become a pathology, as it produces increased imbalance between excitatory and inhibitory circuits, leading to greater susceptibility to further cell loss via calcium-mediated excitotoxicity. Here, we present a mechanistic explanation of how directed brain stimulation might be expected to slow AD progression based on computational simulations in a 470-neuron biomimetic model of a neocortical column. The simulations demonstrate that the addition of low-intensity electrostimulation (neuroprosthesis) to a network undergoing AD-like cell death can raise global activity and break this homeostatic-excitotoxic cascade. The increase in activity within the remaining cells in the column results in lower scaling-driven AMPAR upregulation, reduced imbalances in excitatory and inhibitory circuits, and lower susceptibility to ongoing damage.
细胞死亡和突触功能障碍是 AD 认知能力下降的两个可能原因。随着细胞死亡和突触失去活力,剩余的细胞最初会活动减少。神经元的稳态突触缩放为恢复活动提供了反馈机制。据信,这种稳态机制可以感知细胞内依赖活动的胞质钙水平,并通过增加剩余突触处 AMPA 突触的密度来调整神经元的发射活动,从而达到平衡。缩放机制会增加网络中剩余细胞的发射率,以补偿网络活动的减少。然而,这种效应本身可能成为一种病理学,因为它会在兴奋性和抑制性回路之间产生更大的不平衡,导致通过钙介导的兴奋性毒性进一步增加细胞丢失的易感性。在这里,我们根据一个新皮层柱的 470 个神经元仿生模型的计算模拟,提出了一个关于定向脑刺激如何减缓 AD 进展的机制解释。模拟表明,在经历 AD 样细胞死亡的网络中添加低强度电刺激(神经假体)可以提高全局活动并打破这种稳态-兴奋性毒性级联。柱中剩余细胞内活动的增加导致缩放驱动的 AMPAR 上调减少,兴奋性和抑制性回路的不平衡减少,以及对持续损伤的敏感性降低。