School of Information Engineering, Ningxia University, Yinchuan, 750021, China.
Hefei National Laboratory for Physical Sciences at Microscale, USTC-SJH Joint Center for Human Reproduction and Genetics, School of Life Sciences, University of Science and Technology of China, Hefei, 230027, China.
BMC Bioinformatics. 2020 Jul 23;21(1):331. doi: 10.1186/s12859-020-03665-5.
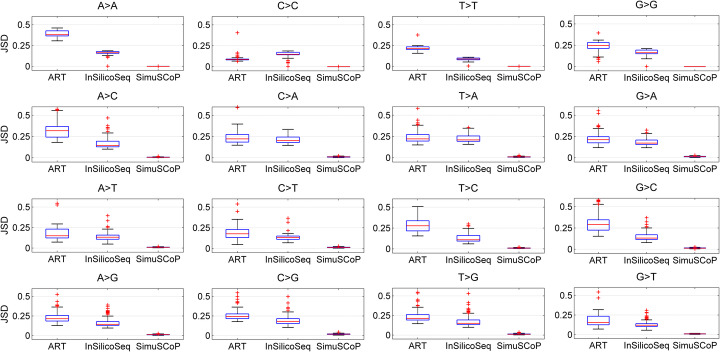
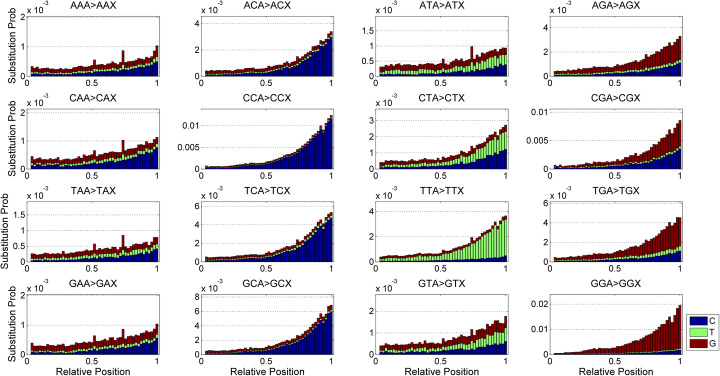
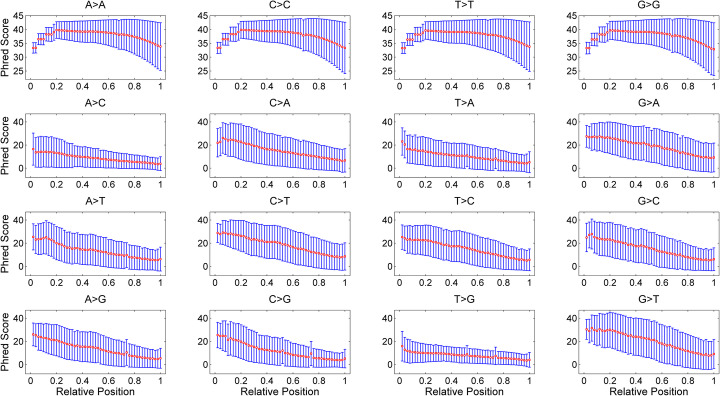
A number of simulators have been developed for emulating next-generation sequencing data by incorporating known errors such as base substitutions and indels. However, their practicality may be degraded by functional and runtime limitations. Particularly, the positional and genomic contextual information is not effectively utilized for reliably characterizing base substitution patterns, as well as the positional and contextual difference of Phred quality scores is not fully investigated. Thus, a more effective and efficient bioinformatics tool is sorely required.
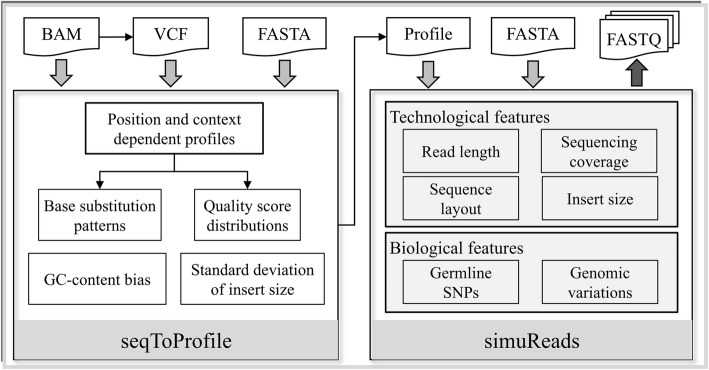
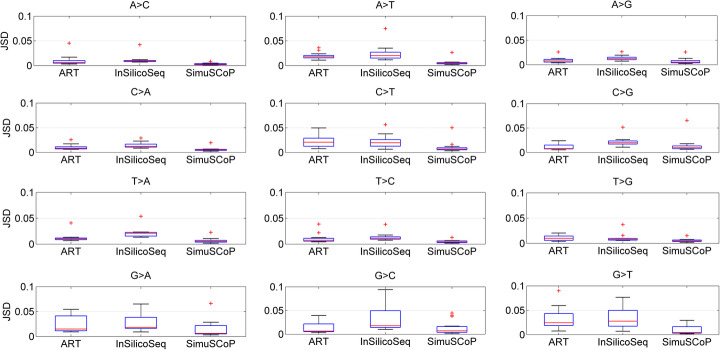
Here, we introduce a novel tool, SimuSCoP, to reliably emulate complex DNA sequencing data. The base substitution patterns and the statistical behavior of quality scores in Illumina sequencing data are fully explored and integrated into the simulation model for reliably emulating datasets for different applications. In addition, an integrated and easy-to-use pipeline is employed in SimuSCoP to facilitate end-to-end simulation of complex samples, and high runtime efficiency is achieved by implementing the tool to run in multithreading with low memory consumption. These features enable SimuSCoP to gets substantial improvements in reliability, functionality, practicality and runtime efficiency. The tool is comprehensively evaluated in multiple aspects including consistency of profiles, simulation of genomic variations and complex tumor samples, and the results demonstrate the advantages of SimuSCoP over existing tools.
SimuSCoP, a new bioinformatics tool is developed to learn informative profiles from real sequencing data and reliably mimic complex data by introducing various genomic variations. We believe that the presented work will catalyse new development of downstream bioinformatics methods for analyzing sequencing data.
已经开发了许多模拟器来模拟下一代测序数据,方法是合并已知的错误,例如碱基替换和插入缺失。然而,它们的实用性可能会因功能和运行时限制而降低。特别是,位置和基因组上下文信息没有有效地用于可靠地描述碱基替换模式,以及 Phred 质量分数的位置和上下文差异也没有得到充分研究。因此,非常需要一种更有效和高效的生物信息学工具。
在这里,我们引入了一种新的工具 SimuSCoP,用于可靠地模拟复杂的 DNA 测序数据。全面探索和整合了 Illumina 测序数据中碱基替换模式和质量分数的统计行为,将其纳入模拟模型中,用于为不同应用可靠地模拟数据集。此外,SimuSCoP 采用了集成且易于使用的流水线,以方便复杂样本的端到端模拟,并通过实现工具以低内存消耗进行多线程运行来实现高运行时效率。这些功能使 SimuSCoP 在可靠性、功能、实用性和运行时效率方面都得到了实质性的提高。该工具在多个方面进行了全面评估,包括分布的一致性、基因组变异和复杂肿瘤样本的模拟,结果表明 SimuSCoP 优于现有工具。
SimuSCoP 是一种新的生物信息学工具,它从真实测序数据中学习信息丰富的分布,并通过引入各种基因组变异来可靠地模拟复杂数据。我们相信,所提出的工作将为分析测序数据的下游生物信息学方法的新发展提供动力。