Walia Rasna R, Xue Li C, Wilkins Katherine, El-Manzalawy Yasser, Dobbs Drena, Honavar Vasant
Bioinformatics and Computational Biology Program, Iowa State University, Ames, Iowa, United States of America; Department of Computer Science, Iowa State University, Ames, Iowa, United States of America.
College of Information Sciences and Technology, Pennsylvania State University, University Park, Pennsylvania, United States of America.
PLoS One. 2014 May 20;9(5):e97725. doi: 10.1371/journal.pone.0097725. eCollection 2014.
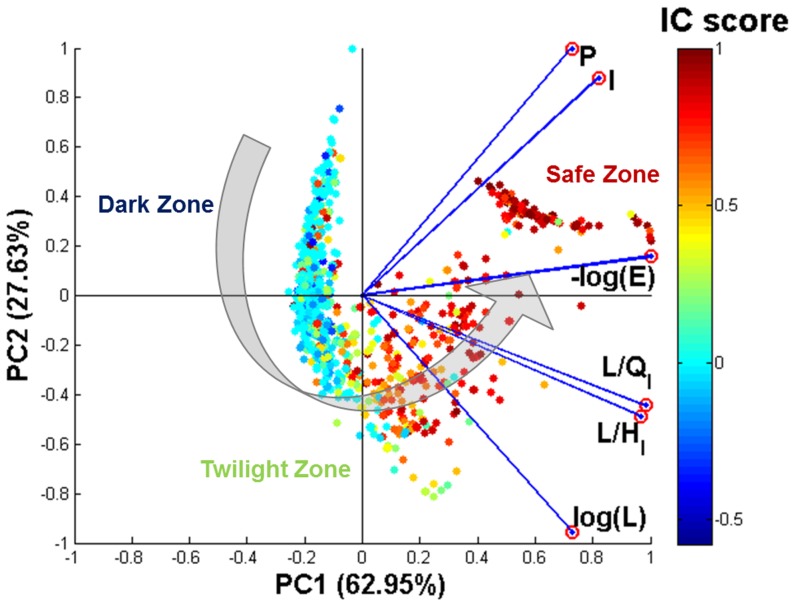
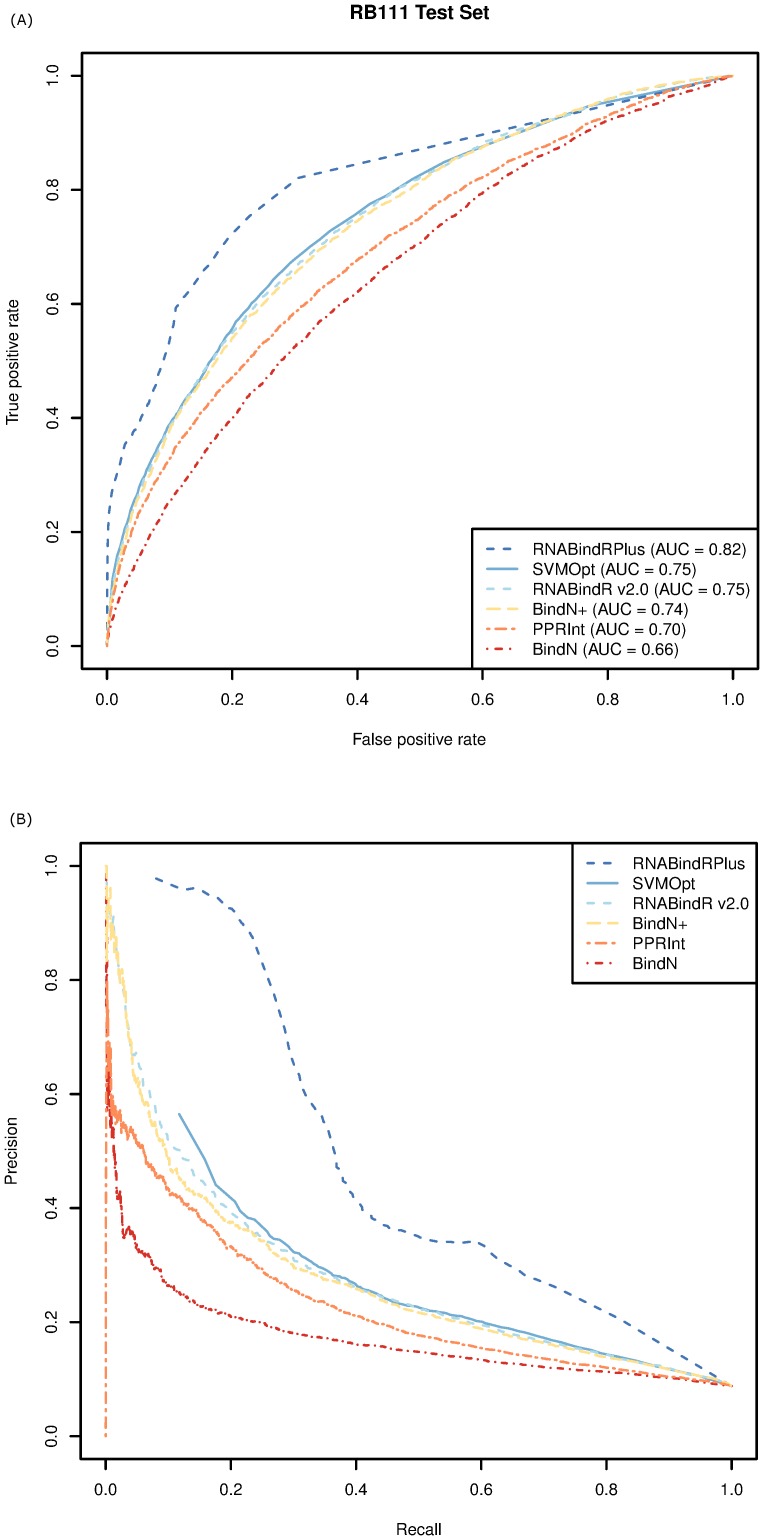
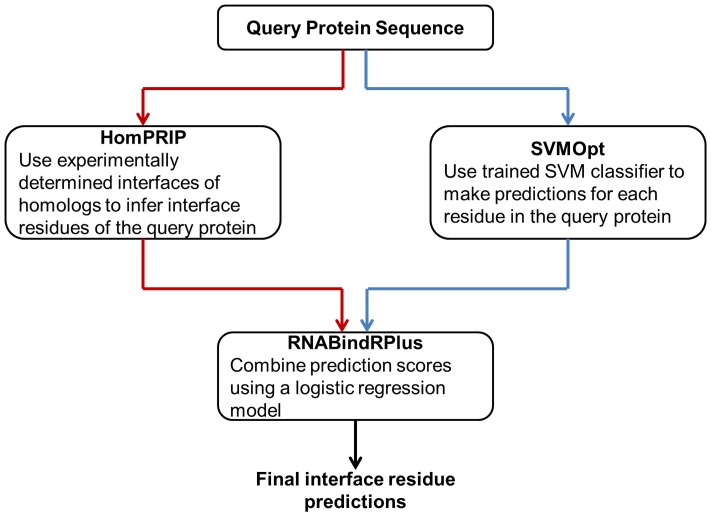
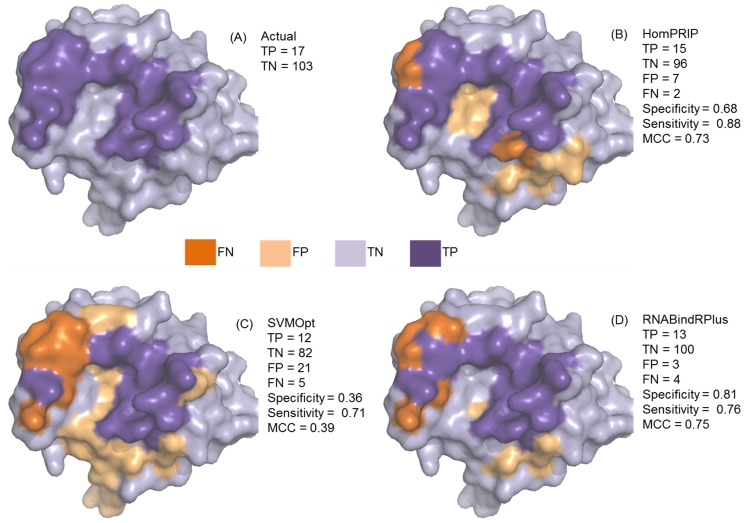
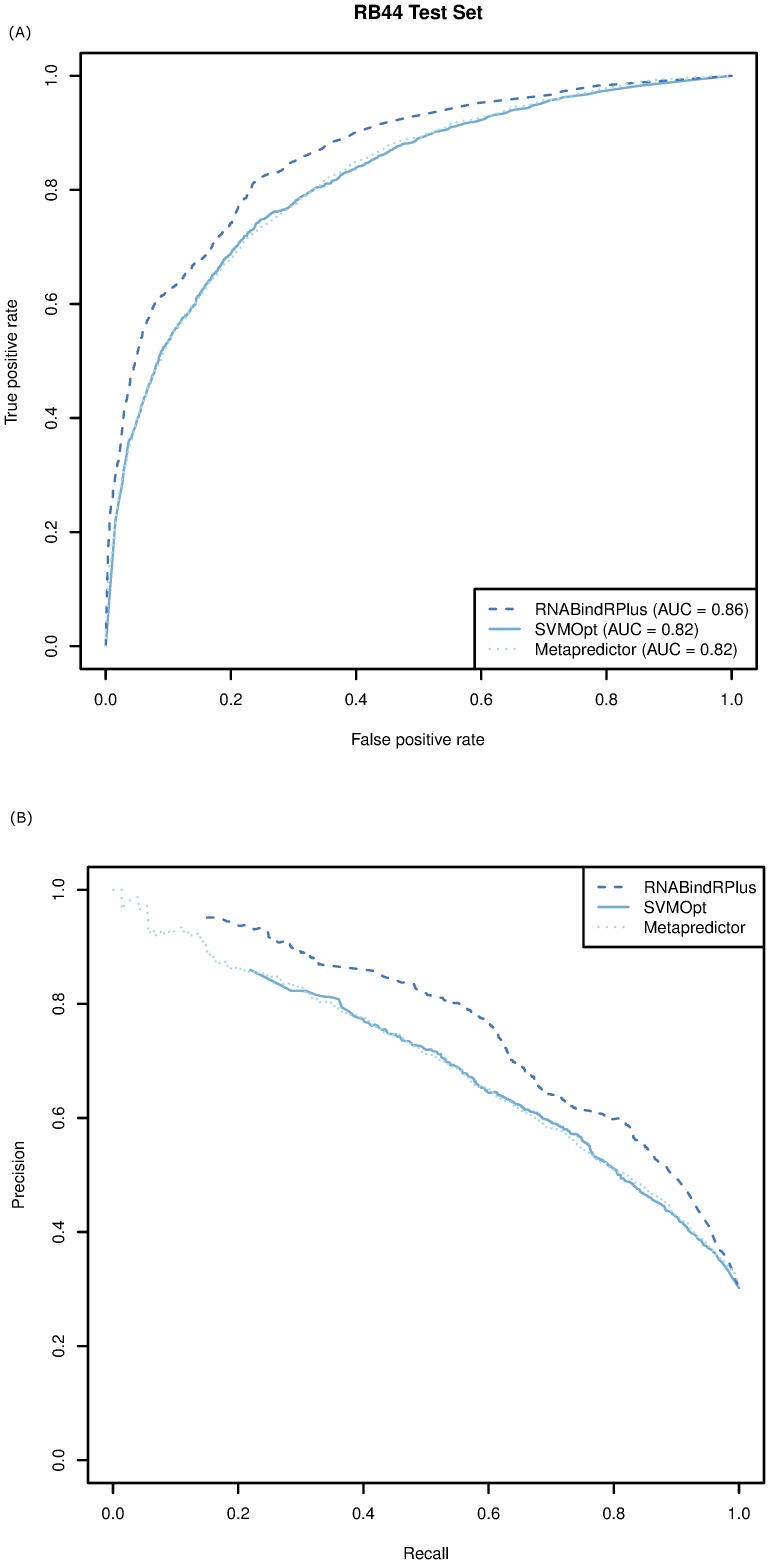
Protein-RNA interactions are central to essential cellular processes such as protein synthesis and regulation of gene expression and play roles in human infectious and genetic diseases. Reliable identification of protein-RNA interfaces is critical for understanding the structural bases and functional implications of such interactions and for developing effective approaches to rational drug design. Sequence-based computational methods offer a viable, cost-effective way to identify putative RNA-binding residues in RNA-binding proteins. Here we report two novel approaches: (i) HomPRIP, a sequence homology-based method for predicting RNA-binding sites in proteins; (ii) RNABindRPlus, a new method that combines predictions from HomPRIP with those from an optimized Support Vector Machine (SVM) classifier trained on a benchmark dataset of 198 RNA-binding proteins. Although highly reliable, HomPRIP cannot make predictions for the unaligned parts of query proteins and its coverage is limited by the availability of close sequence homologs of the query protein with experimentally determined RNA-binding sites. RNABindRPlus overcomes these limitations. We compared the performance of HomPRIP and RNABindRPlus with that of several state-of-the-art predictors on two test sets, RB44 and RB111. On a subset of proteins for which homologs with experimentally determined interfaces could be reliably identified, HomPRIP outperformed all other methods achieving an MCC of 0.63 on RB44 and 0.83 on RB111. RNABindRPlus was able to predict RNA-binding residues of all proteins in both test sets, achieving an MCC of 0.55 and 0.37, respectively, and outperforming all other methods, including those that make use of structure-derived features of proteins. More importantly, RNABindRPlus outperforms all other methods for any choice of tradeoff between precision and recall. An important advantage of both HomPRIP and RNABindRPlus is that they rely on readily available sequence and sequence-derived features of RNA-binding proteins. A webserver implementation of both methods is freely available at http://einstein.cs.iastate.edu/RNABindRPlus/.
蛋白质与RNA的相互作用对于蛋白质合成和基因表达调控等基本细胞过程至关重要,并在人类感染性疾病和遗传性疾病中发挥作用。可靠地识别蛋白质-RNA界面对于理解此类相互作用的结构基础和功能影响以及开发合理药物设计的有效方法至关重要。基于序列的计算方法为识别RNA结合蛋白中假定的RNA结合残基提供了一种可行且具有成本效益的方法。在此,我们报告两种新方法:(i)HomPRIP,一种基于序列同源性预测蛋白质中RNA结合位点的方法;(ii)RNABindRPlus,一种将HomPRIP的预测结果与基于198个RNA结合蛋白的基准数据集训练的优化支持向量机(SVM)分类器的预测结果相结合的新方法。尽管HomPRIP非常可靠,但它无法对查询蛋白未比对的部分进行预测,其覆盖范围受到具有实验确定的RNA结合位点的查询蛋白的紧密序列同源物可用性的限制。RNABindRPlus克服了这些限制。我们在两个测试集RB44和RB111上比较了HomPRIP和RNABindRPlus与几种最先进预测器的性能。在能够可靠识别具有实验确定界面的同源物的一部分蛋白质上,HomPRIP优于所有其他方法,在RB44上的马修斯相关系数(MCC)为0.63,在RB111上为0.83。RNABindRPlus能够预测两个测试集中所有蛋白质的RNA结合残基,MCC分别为0.55和0.37,并且优于所有其他方法,包括那些利用蛋白质结构衍生特征的方法。更重要的是,对于精度和召回率之间的任何权衡选择,RNABindRPlus都优于所有其他方法。HomPRIP和RNABindRPlus的一个重要优点是它们依赖于RNA结合蛋白易于获得的序列和序列衍生特征。这两种方法的网络服务器实现可在http://einstein.cs.iastate.edu/RNABindRPlus/免费获取。