Department of physiology, School of Medicine, Chungnam National University, Daejeon, Republic of Korea.
Department of Endocrinology, Chungnam National University Hospital, Daejeon, Republic of Korea.
PLoS One. 2014 Jun 6;9(6):e98670. doi: 10.1371/journal.pone.0098670. eCollection 2014.
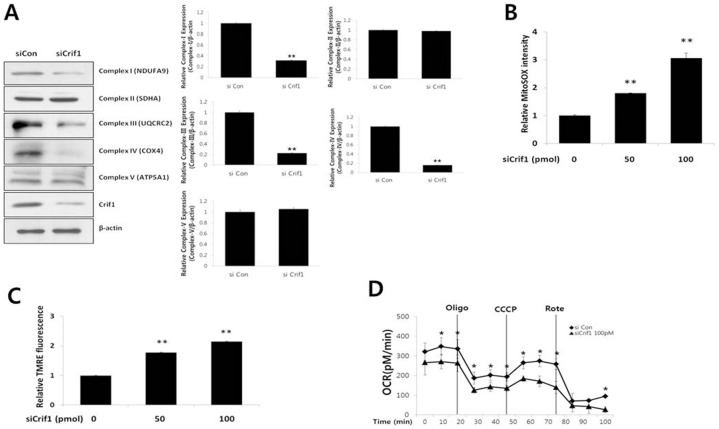
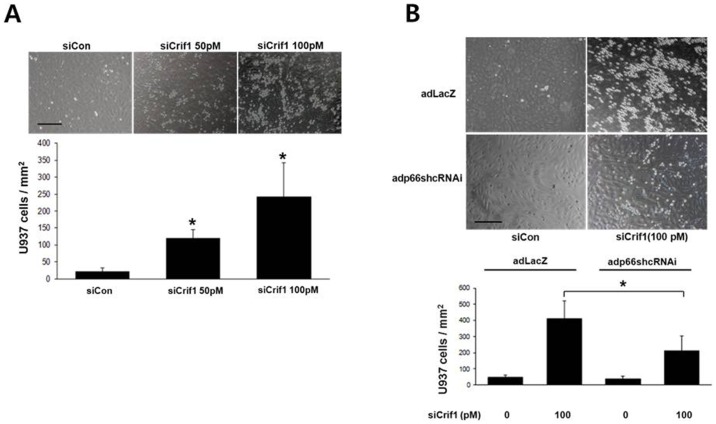
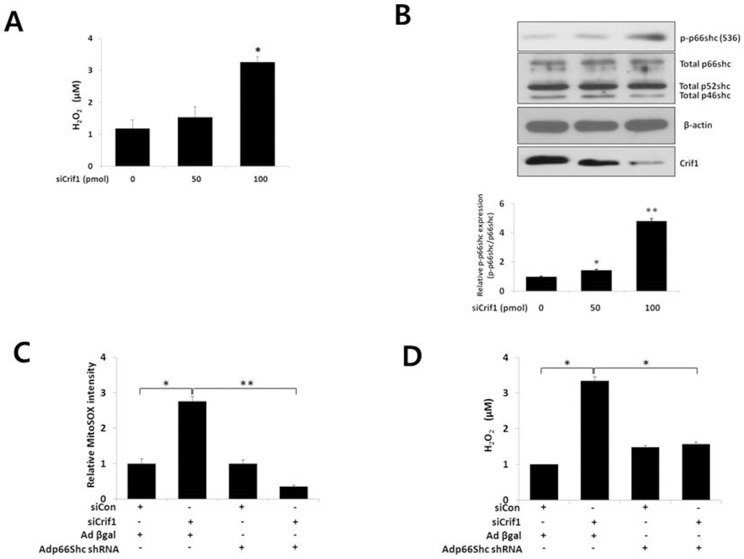
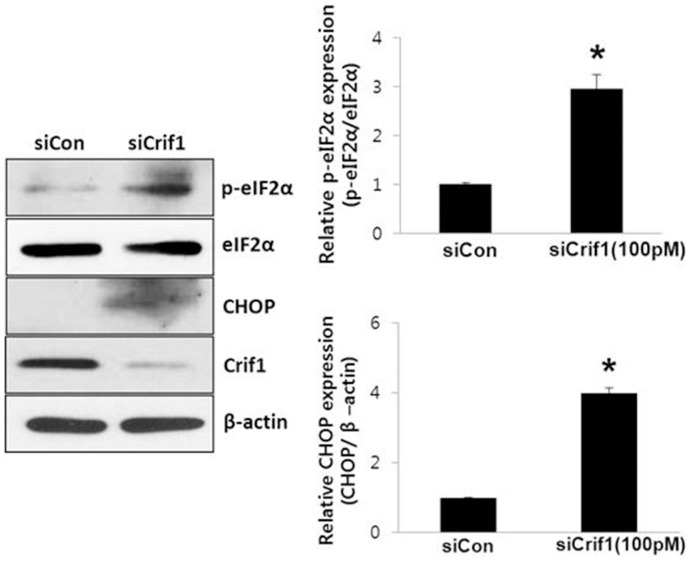
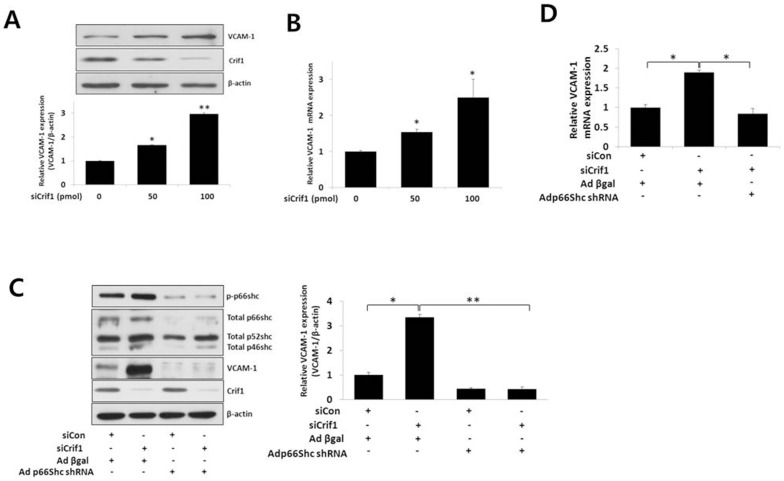
Mitochondrial dysfunction has been implicated in the pathophysiology of various cardiovascular diseases. CRIF1 is a protein present in the mitochondria associated with large mitoribosomal subunits, and CRIF1 knockdown induces mitochondrial dysfunction and promotes ROS production. p66shc is a redox enzyme implicated in mitochondrial ROS generation and translation of oxidative signals and, therefore, is a key factor for oxidative stress in endothelial cells. In this study, we investigated whether mitochondrial dysfunction induced by CRIF1 knockdown induces p66shc stimulation and plays any role in mitochondrial dysfunction-induced endothelial activation. Knockdown of CRIF1 decreased the expression of mitochondrial oxidative phosphorylation (OXPHOS) complexes I, III and IV, leading to increased mitochondrial ROS (mtROS) and hyperpolarization of the mitochondrial membrane potential. Knockdown of CRIF1 also stimulated phosphorylation of p66shc and increased cytosolic ROS in endothelial cells. Furthermore, the expression of vascular cell adhesion molecule-1 and endoplasmic reticulum stress proteins were increased upon CRIF1 knockdown in endothelial cells. However, p66shc knockdown blunted the alteration in mitochondrial dynamics and ROS production in CRIF1 knockdown endothelial cells. In addition, p66shc knockdown reduced the CRIF1 knockdown-induced increases in adhesion between monocytes and endothelial cells. Taken together, these results suggest that CRIF1 knockdown partially induces endothelial activation via increased ROS production and phosphorylation of p66shc.
线粒体功能障碍与各种心血管疾病的病理生理学有关。CRIF1 是一种存在于与大型线粒体核糖体亚基相关的线粒体中的蛋白质,CRIF1 敲低会诱导线粒体功能障碍并促进 ROS 产生。p66shc 是一种与线粒体 ROS 生成和氧化信号转译有关的氧化还原酶,因此是内皮细胞氧化应激的关键因素。在这项研究中,我们研究了 CRIF1 敲低诱导的线粒体功能障碍是否会诱导 p66shc 刺激,并在诱导的线粒体功能障碍的内皮细胞激活中发挥任何作用。CRIF1 的敲低降低了线粒体氧化磷酸化 (OXPHOS) 复合物 I、III 和 IV 的表达,导致线粒体 ROS (mtROS) 增加和线粒体膜电位去极化。CRIF1 的敲低还刺激了 p66shc 的磷酸化,并增加了内皮细胞中的细胞质 ROS。此外,在内皮细胞中 CRIF1 敲低后,血管细胞黏附分子-1 和内质网应激蛋白的表达增加。然而,p66shc 的敲低削弱了 CRIF1 敲低内皮细胞中线粒体动力学和 ROS 产生的改变。此外,p66shc 的敲低减少了 CRIF1 敲低诱导的单核细胞与内皮细胞之间的黏附增加。总之,这些结果表明,CRIF1 敲低部分通过增加 ROS 产生和 p66shc 的磷酸化诱导内皮细胞激活。