Kizilkaya Kadir, Fernando Rohan L, Garrick Dorian J
Department of Animal Science, Iowa State University, Ames IA 50011, USA.
Genet Sel Evol. 2014 Jun 9;46(1):37. doi: 10.1186/1297-9686-46-37.
Accuracy of genomic prediction depends on number of records in the training population, heritability, effective population size, genetic architecture, and relatedness of training and validation populations. Many traits have ordered categories including reproductive performance and susceptibility or resistance to disease. Categorical scores are often recorded because they are easier to obtain than continuous observations. Bayesian linear regression has been extended to the threshold model for genomic prediction. The objective of this study was to quantify reductions in accuracy for ordinal categorical traits relative to continuous traits.
Efficiency of genomic prediction was evaluated for heritabilities of 0.10, 0.25 or 0.50. Phenotypes were simulated for 2250 purebred animals using 50 QTL selected from actual 50k SNP (single nucleotide polymorphism) genotypes giving a proportion of causal to total loci of.0001. A Bayes C π threshold model simultaneously fitted all 50k markers except those that represented QTL. Estimated SNP effects were utilized to predict genomic breeding values in purebred (n = 239) or multibreed (n = 924) validation populations. Correlations between true and predicted genomic merit in validation populations were used to assess predictive ability.
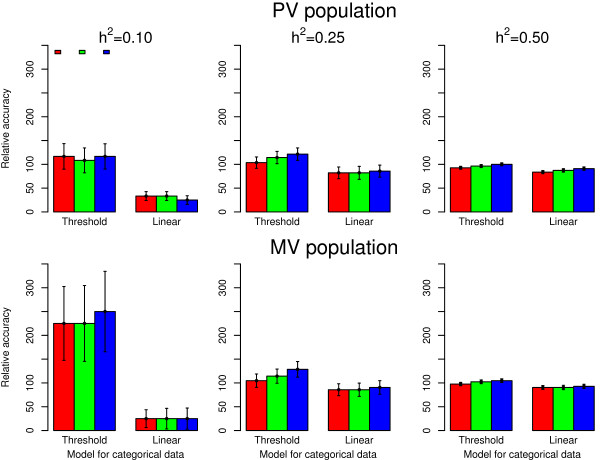
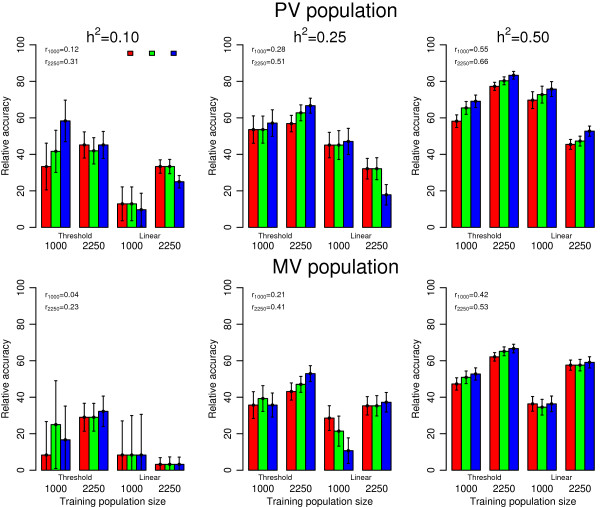
Accuracies of genomic estimated breeding values ranged from 0.12 to 0.66 for purebred and from 0.04 to 0.53 for multibreed validation populations based on Bayes C π linear model analysis of the simulated underlying variable. Accuracies for ordinal categorical scores analyzed by the Bayes C π threshold model were 20% to 50% lower and ranged from 0.04 to 0.55 for purebred and from 0.01 to 0.44 for multibreed validation populations. Analysis of ordinal categorical scores using a linear model resulted in further reductions in accuracy.
Threshold traits result in markedly lower accuracy than a linear model on the underlying variable. To achieve an accuracy equal or greater than for continuous phenotypes with a training population of 1000 animals, a 2.25 fold increase in training population size was required for categorical scores fitted with the threshold model. The threshold model resulted in higher accuracies than the linear model and its advantage was greatest when training populations were smallest.
基因组预测的准确性取决于训练群体中的记录数量、遗传力、有效群体大小、遗传结构以及训练群体与验证群体的亲缘关系。许多性状具有有序类别,包括繁殖性能以及对疾病的易感性或抗性。通常记录分类得分,因为它们比连续观测值更容易获得。贝叶斯线性回归已扩展到用于基因组预测的阈值模型。本研究的目的是量化序数分类性状相对于连续性状准确性的降低程度。
针对遗传力为0.10、0.25或0.50的情况评估基因组预测的效率。使用从实际50k单核苷酸多态性(SNP)基因型中选择的50个数量性状位点(QTL)为2250只纯种动物模拟表型,使得因果位点与总位点的比例为0.0001。贝叶斯Cπ阈值模型同时拟合除代表QTL的那些标记之外的所有50k个标记。利用估计的SNP效应在纯种(n = 239)或多品种(n = 924)验证群体中预测基因组育种值。验证群体中真实和预测的基因组优点之间的相关性用于评估预测能力。
基于对模拟潜在变量的贝叶斯Cπ线性模型分析,纯种验证群体的基因组估计育种值准确性范围为0.12至0.66,多品种验证群体为0.04至0.53。通过贝叶斯Cπ阈值模型分析的序数分类得分的准确性低20%至50%,纯种验证群体范围为0.04至0.55,多品种验证群体为0.01至0.44。使用线性模型分析序数分类得分导致准确性进一步降低。
阈值性状导致的准确性明显低于基于潜在变量的线性模型。为了在训练群体为1000只动物的情况下实现与连续表型相等或更高的准确性,对于采用阈值模型拟合的分类得分,训练群体大小需要增加2.25倍。阈值模型比线性模型产生更高的准确性,并且当训练群体最小时其优势最大。