Lindemose Søren, Jensen Michael K, Van de Velde Jan, O'Shea Charlotte, Heyndrickx Ken S, Workman Christopher T, Vandepoele Klaas, Skriver Karen, De Masi Federico
Department of Biology, University of Copenhagen, 2200 Copenhagen, Denmark.
Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, DK-2970 Hørsholm, Denmark.
Nucleic Acids Res. 2014 Jul;42(12):7681-93. doi: 10.1093/nar/gku502. Epub 2014 Jun 9.
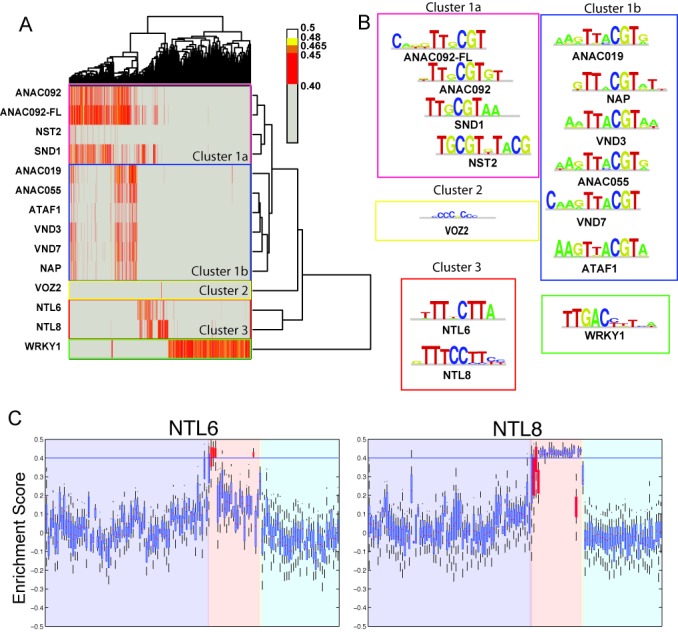
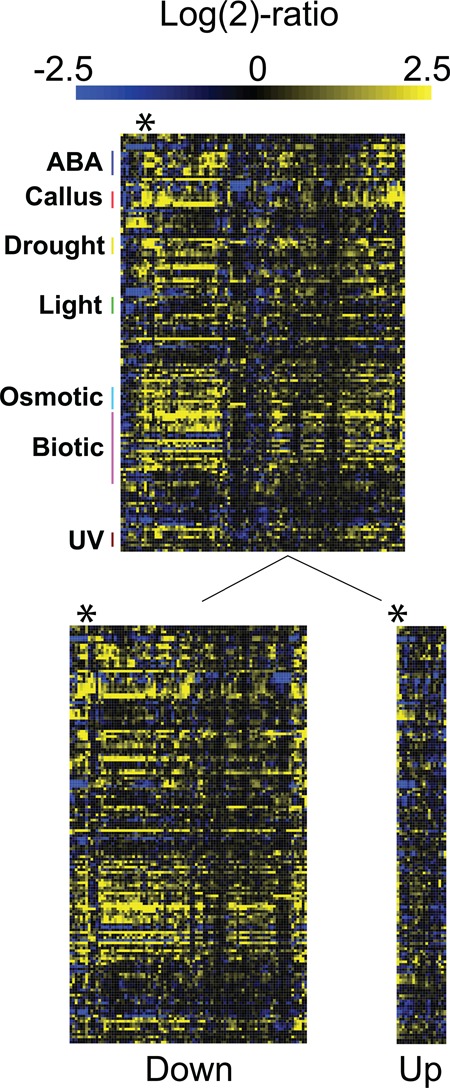
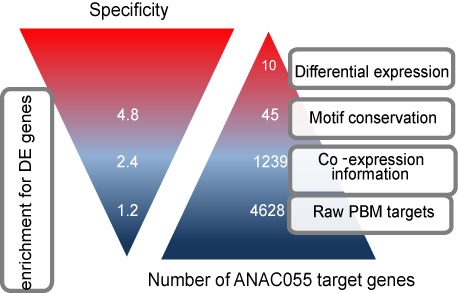
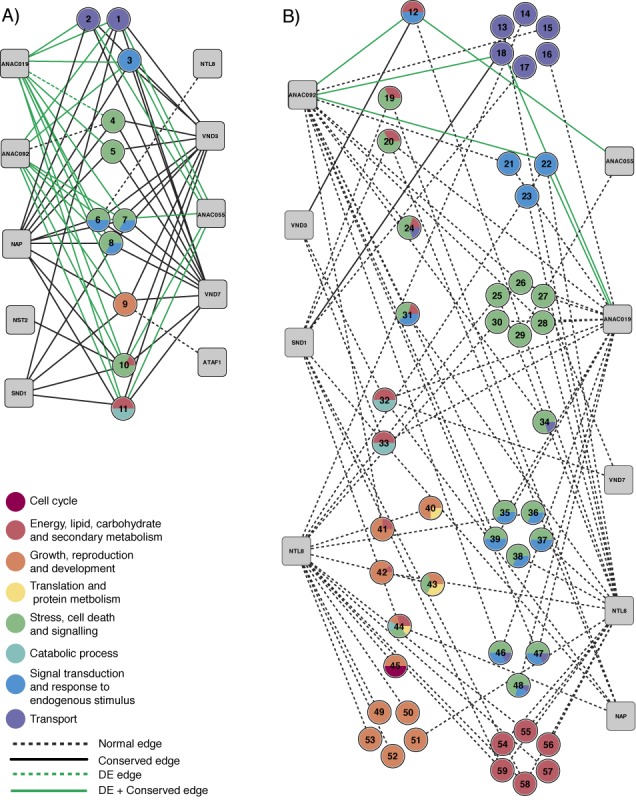
Target gene identification for transcription factors is a prerequisite for the systems wide understanding of organismal behaviour. NAM-ATAF1/2-CUC2 (NAC) transcription factors are amongst the largest transcription factor families in plants, yet limited data exist from unbiased approaches to resolve the DNA-binding preferences of individual members. Here, we present a TF-target gene identification workflow based on the integration of novel protein binding microarray data with gene expression and multi-species promoter sequence conservation to identify the DNA-binding specificities and the gene regulatory networks of 12 NAC transcription factors. Our data offer specific single-base resolution fingerprints for most TFs studied and indicate that NAC DNA-binding specificities might be predicted from their DNA-binding domain's sequence. The developed methodology, including the application of complementary functional genomics filters, makes it possible to translate, for each TF, protein binding microarray data into a set of high-quality target genes. With this approach, we confirm NAC target genes reported from independent in vivo analyses. We emphasize that candidate target gene sets together with the workflow associated with functional modules offer a strong resource to unravel the regulatory potential of NAC genes and that this workflow could be used to study other families of transcription factors.
转录因子的靶基因鉴定是全面了解生物体行为的先决条件。NAM-ATAF1/2-CUC2(NAC)转录因子是植物中最大的转录因子家族之一,但通过无偏差方法解析单个成员的DNA结合偏好的数据有限。在此,我们提出了一种转录因子-靶基因鉴定工作流程,该流程基于将新的蛋白质结合微阵列数据与基因表达和多物种启动子序列保守性相结合,以鉴定12个NAC转录因子的DNA结合特异性和基因调控网络。我们的数据为大多数研究的转录因子提供了特定的单碱基分辨率指纹,并表明NAC的DNA结合特异性可能可以从其DNA结合结构域的序列中预测出来。所开发的方法,包括应用互补的功能基因组学筛选,使得能够将每个转录因子的蛋白质结合微阵列数据转化为一组高质量的靶基因。通过这种方法,我们证实了独立体内分析报告的NAC靶基因。我们强调,候选靶基因集以及与功能模块相关的工作流程为揭示NAC基因的调控潜力提供了强大的资源,并且该工作流程可用于研究其他转录因子家族。