Daly Rónán, Rogers Simon, Wandy Joe, Jankevics Andris, Burgess Karl E V, Breitling Rainer
School of Computing Science, University of Glasgow, Glasgow, Manchester Institute of Biotechnology, Faculty of Life Sciences, University of Manchester, Manchester and Institute of Infection, Immunity and Inflammation, University of Glasgow, Glasgow, UK.
Bioinformatics. 2014 Oct;30(19):2764-71. doi: 10.1093/bioinformatics/btu370. Epub 2014 Jun 9.
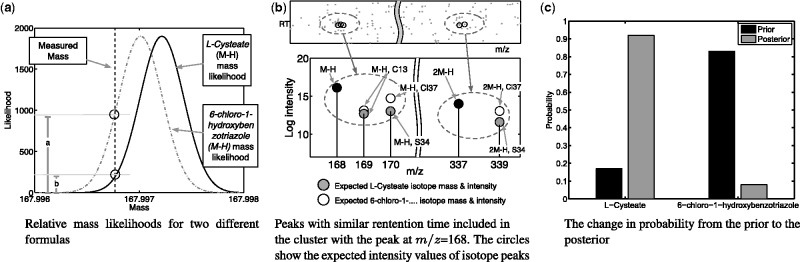
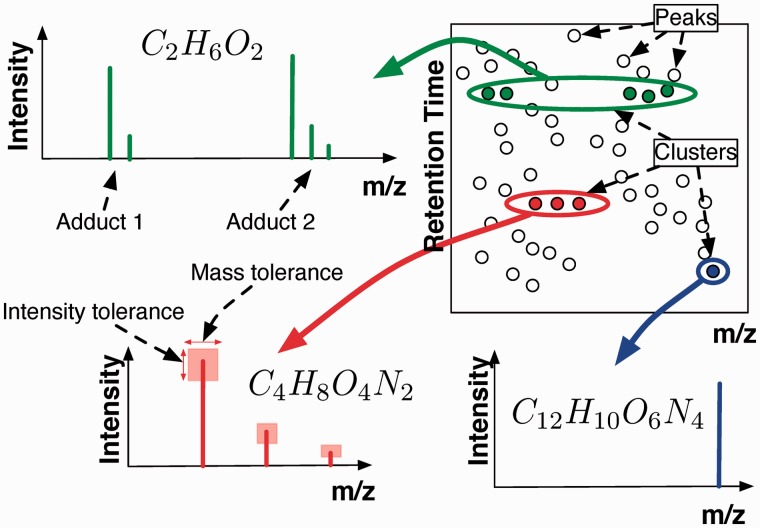
The use of liquid chromatography coupled to mass spectrometry has enabled the high-throughput profiling of the metabolite composition of biological samples. However, the large amount of data obtained can be difficult to analyse and often requires computational processing to understand which metabolites are present in a sample. This article looks at the dual problem of annotating peaks in a sample with a metabolite, together with putatively annotating whether a metabolite is present in the sample. The starting point of the approach is a Bayesian clustering of peaks into groups, each corresponding to putative adducts and isotopes of a single metabolite.
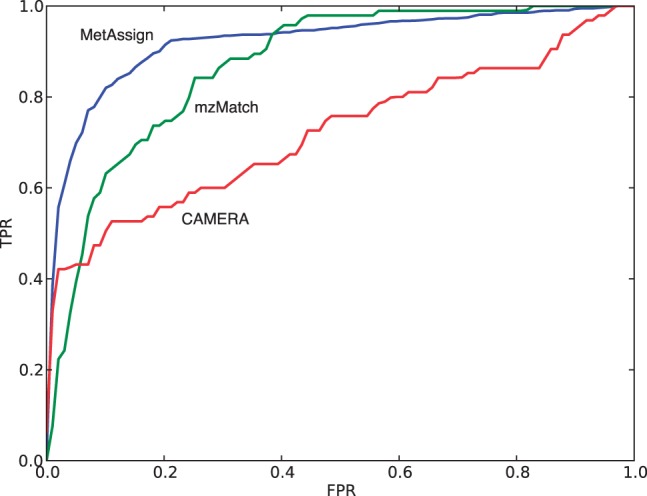
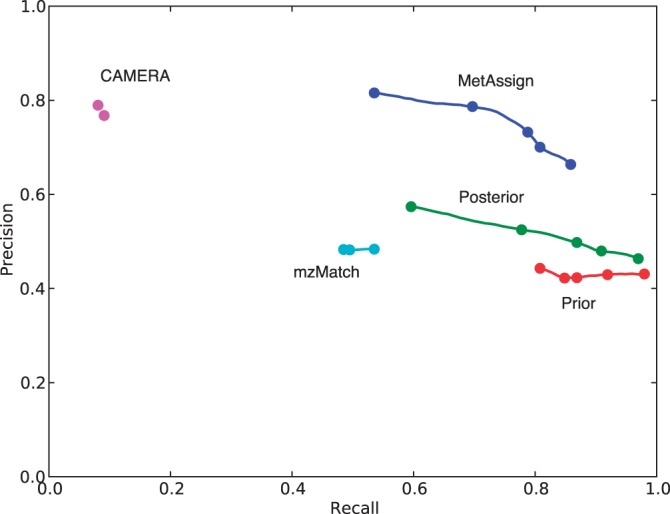
The Bayesian modelling introduced here combines information from the mass-to-charge ratio, retention time and intensity of each peak, together with a model of the inter-peak dependency structure, to increase the accuracy of peak annotation. The results inherently contain a quantitative estimate of confidence in the peak annotations and allow an accurate trade-off between precision and recall. Extensive validation experiments using authentic chemical standards show that this system is able to produce more accurate putative identifications than other state-of-the-art systems, while at the same time giving a probabilistic measure of confidence in the annotations.
The software has been implemented as part of the mzMatch metabolomics analysis pipeline, which is available for download at http://mzmatch.sourceforge.net/.
液相色谱-质谱联用技术能够对生物样品的代谢物组成进行高通量分析。然而,所获得的大量数据可能难以分析,通常需要进行计算处理才能了解样品中存在哪些代谢物。本文探讨了用代谢物对样品中的峰进行注释以及推测样品中是否存在某种代谢物这一双重问题。该方法的出发点是将峰进行贝叶斯聚类,分为不同的组,每组对应一种代谢物的假定加合物和同位素。
本文介绍的贝叶斯模型结合了每个峰的质荷比、保留时间和强度信息,以及峰间依赖结构模型,以提高峰注释的准确性。结果中固有地包含了对峰注释置信度的定量估计,并允许在精度和召回率之间进行准确权衡。使用真实化学标准品进行的广泛验证实验表明,该系统能够比其他现有技术系统产生更准确的假定鉴定结果,同时还能给出注释置信度的概率度量。
该软件已作为mzMatch代谢组学分析管道的一部分实现,可从http://mzmatch.sourceforge.net/下载。