Department of Cellular and Molecular Medicine, Arizona Health Sciences Center, The University of Arizona, Tucson, Arizona 85735, USA.
BMC Med Genomics. 2014 Jun 10;7:33. doi: 10.1186/1755-8794-7-33.
Numerous microarray-based prognostic gene expression signatures of primary neoplasms have been published but often with little concurrence between studies, thus limiting their clinical utility. We describe a methodology using logistic regression, which circumvents limitations of conventional Kaplan Meier analysis. We applied this approach to a thrice-analyzed and published squamous cell carcinoma (SQCC) of the lung data set, with the objective of identifying gene expressions predictive of early death versus long survival in early-stage disease. A similar analysis was applied to a data set of triple negative breast carcinoma cases, which present similar clinical challenges.
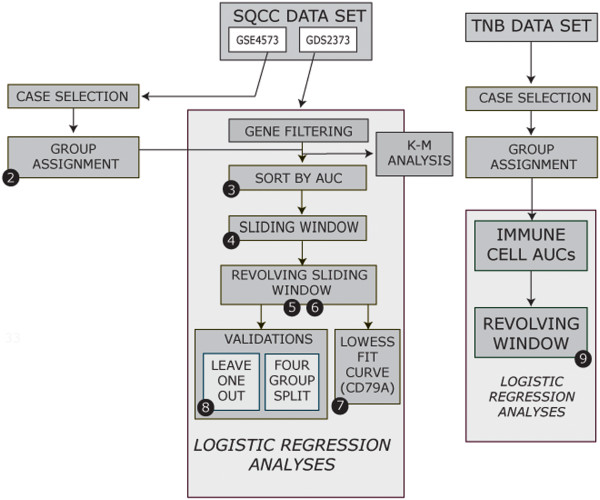
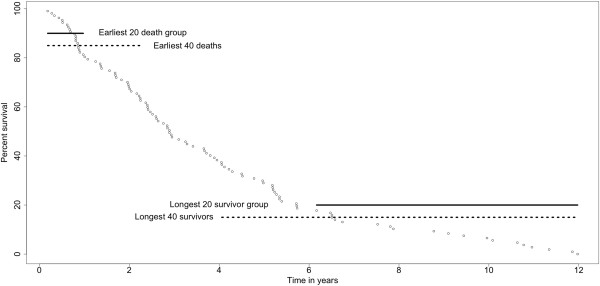
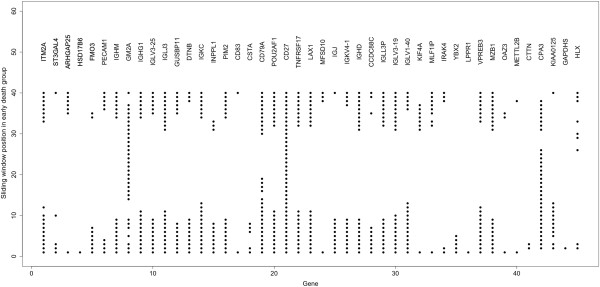
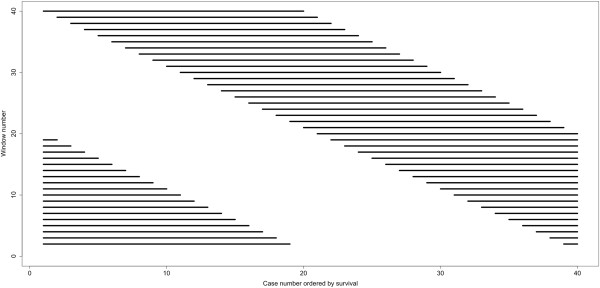
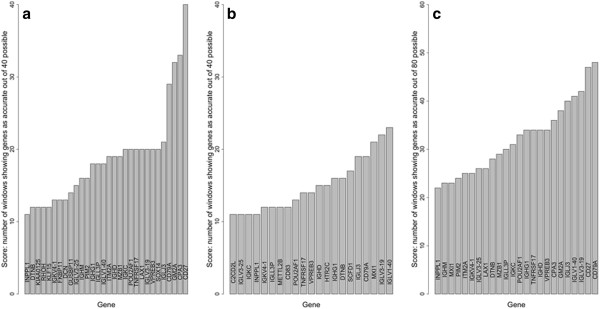
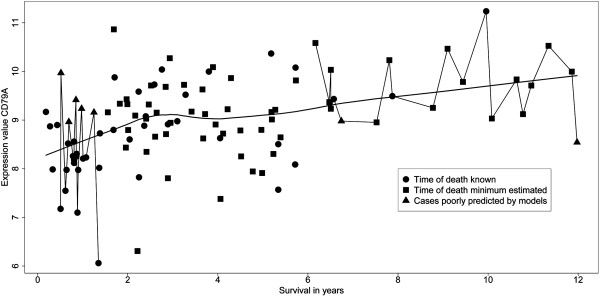
Important to our approach is the selection of homogenous patient groups for comparison. In the lung study, we selected two groups (including only stages I and II), equal in size, of earliest deaths and longest survivors. Genes varying at least four-fold were tested by logistic regression for accuracy of prediction (area under a ROC plot). The gene list was refined by applying two sliding-window analyses and by validations using a leave-one-out approach and model building with validation subsets. In the breast study, a similar logistic regression analysis was used after selecting appropriate cases for comparison.
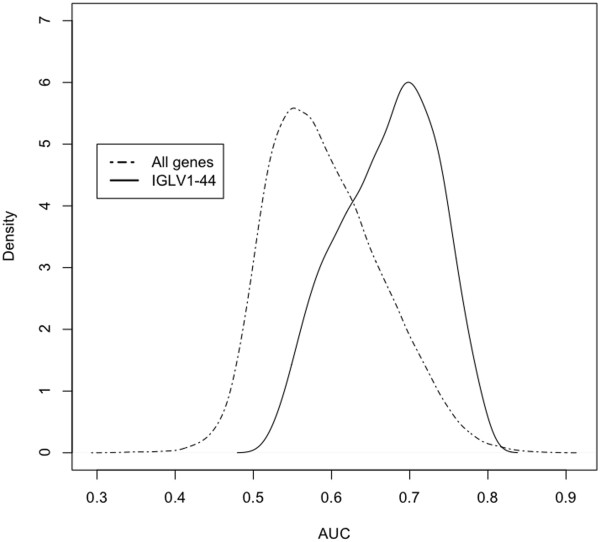
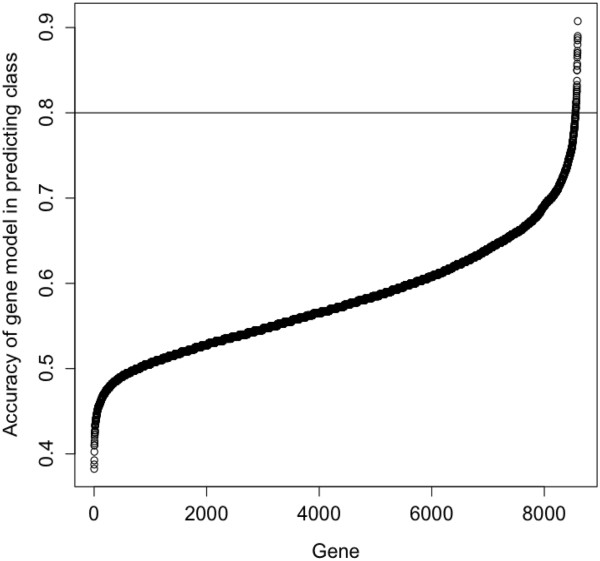
A total of 8594 variable genes were tested for accuracy in predicting earliest deaths versus longest survivors in SQCC. After applying the two sliding window and the leave-one-out analyses, 24 prognostic genes were identified; most of them were B-cell related. When the same data set of stage I and II cases was analyzed using a conventional Kaplan Meier (KM) approach, we identified fewer immune-related genes among the most statistically significant hits; when stage III cases were included, most of the prognostic genes were missed. Interestingly, logistic regression analysis of the breast cancer data set identified many immune-related genes predictive of clinical outcome.
Stratification of cases based on clinical data, careful selection of two groups for comparison, and the application of logistic regression analysis substantially improved predictive accuracy in comparison to conventional KM approaches. B cell-related genes dominated the list of prognostic genes in early stage SQCC of the lung and triple negative breast cancer.
已发表了许多基于微阵列的原发性肿瘤预后基因表达特征,但这些研究之间往往一致性较差,因此限制了其临床应用。我们描述了一种使用逻辑回归的方法,该方法规避了传统的 Kaplan-Meier 分析的局限性。我们将这种方法应用于已分析并发表的三次鳞状细胞肺癌(SQCC)数据集中,目的是确定预测早期疾病中早期死亡与长期生存的基因表达。对三阴性乳腺癌病例数据集进行了类似的分析,这些病例具有相似的临床挑战。
我们方法的重要之处在于选择用于比较的同质患者组。在肺癌研究中,我们选择了两个大小相等的最早死亡和最长生存的组(仅包括 I 期和 II 期)。通过逻辑回归对数个差异至少四倍的基因进行准确性测试(ROC 图下面积)。通过应用两个滑动窗口分析和使用留一法验证以及使用验证子集进行模型构建来对基因列表进行精炼。在乳腺癌研究中,在选择适当的病例进行比较后,使用类似的逻辑回归分析。
总共对 8594 个变量基因进行了测试,以预测 SQCC 中最早死亡与最长生存的准确性。应用两个滑动窗口和留一法分析后,确定了 24 个预后基因;其中大多数与 B 细胞有关。当使用传统的 Kaplan-Meier(KM)方法分析 I 期和 II 期病例的相同数据集时,我们在最具统计学意义的命中者中发现了较少的免疫相关基因;当包括 III 期病例时,大多数预后基因都被遗漏了。有趣的是,对乳腺癌数据集的逻辑回归分析确定了许多预测临床结果的免疫相关基因。
基于临床数据对病例进行分层,仔细选择两组进行比较,并应用逻辑回归分析,与传统的 KM 方法相比,大大提高了预测准确性。在早期 SQCC 和三阴性乳腺癌中,B 细胞相关基因主导了预后基因列表。