Kukat Alexandra, Dogan Sukru Anil, Edgar Daniel, Mourier Arnaud, Jacoby Christoph, Maiti Priyanka, Mauer Jan, Becker Christina, Senft Katharina, Wibom Rolf, Kudin Alexei P, Hultenby Kjell, Flögel Ulrich, Rosenkranz Stephan, Ricquier Daniel, Kunz Wolfram S, Trifunovic Aleksandra
Cologne Excellence Cluster on Cellular Stress Responses in Aging-Associated Diseases (CECAD) and Institute for Mitochondrial Diseases and Aging, Medical Faculty, University of Cologne, Cologne, Germany; Department of Laboratory Medicine, Karolinska Institutet, Stockholm, Sweden.
Cologne Excellence Cluster on Cellular Stress Responses in Aging-Associated Diseases (CECAD) and Institute for Mitochondrial Diseases and Aging, Medical Faculty, University of Cologne, Cologne, Germany.
PLoS Genet. 2014 Jun 19;10(6):e1004385. doi: 10.1371/journal.pgen.1004385. eCollection 2014 Jun.
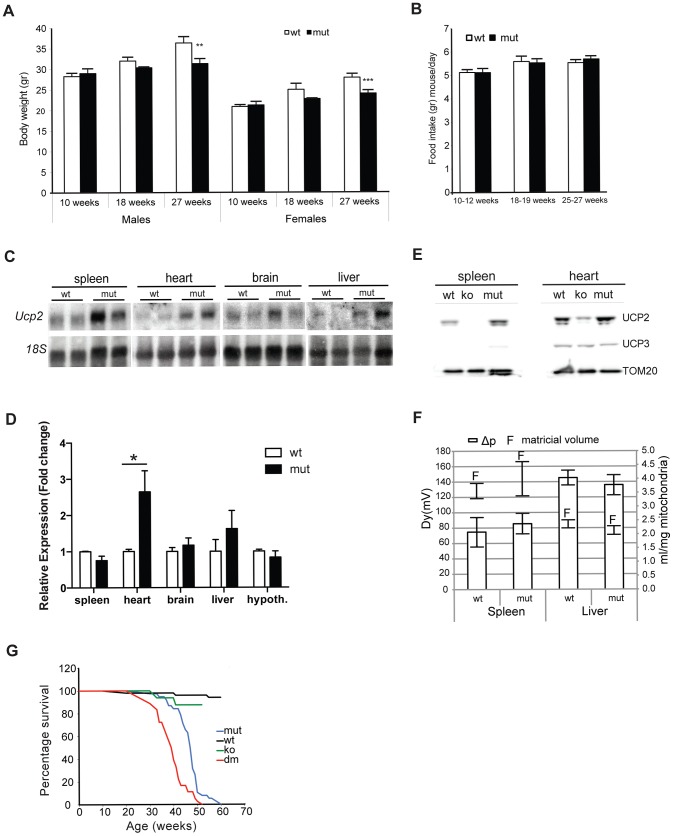
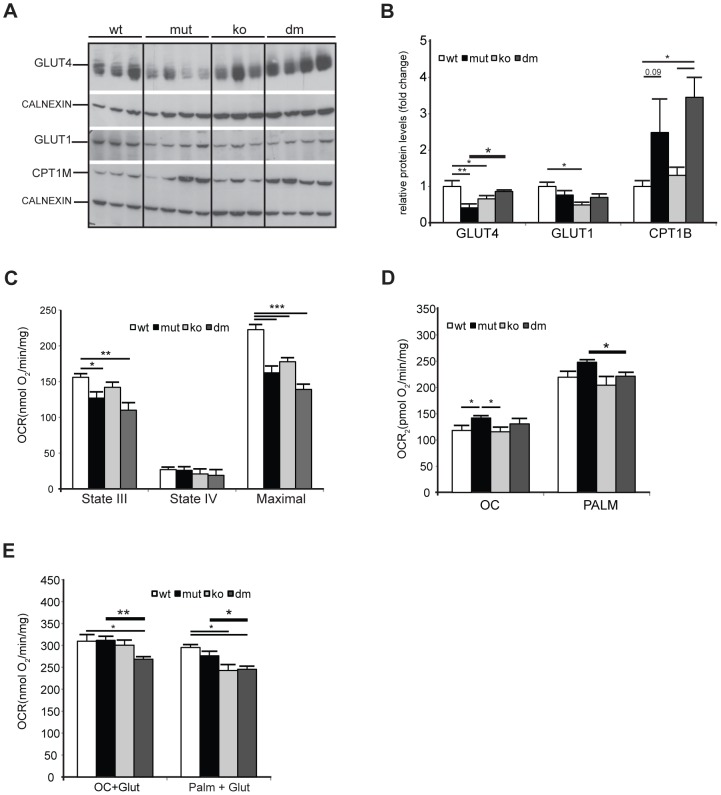
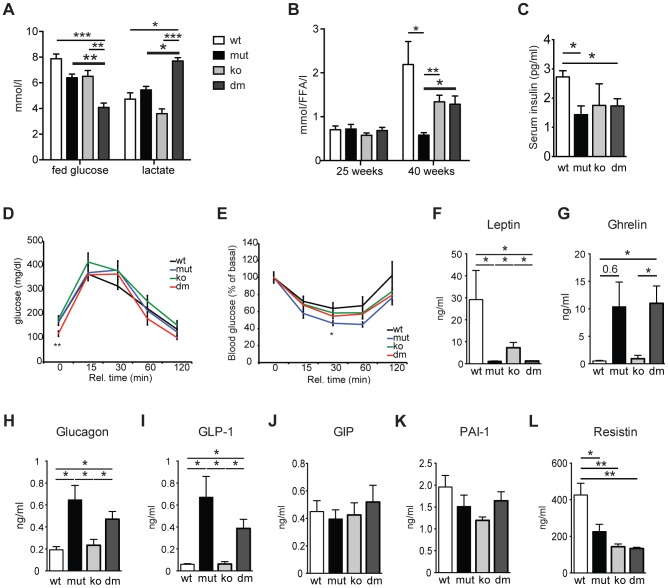
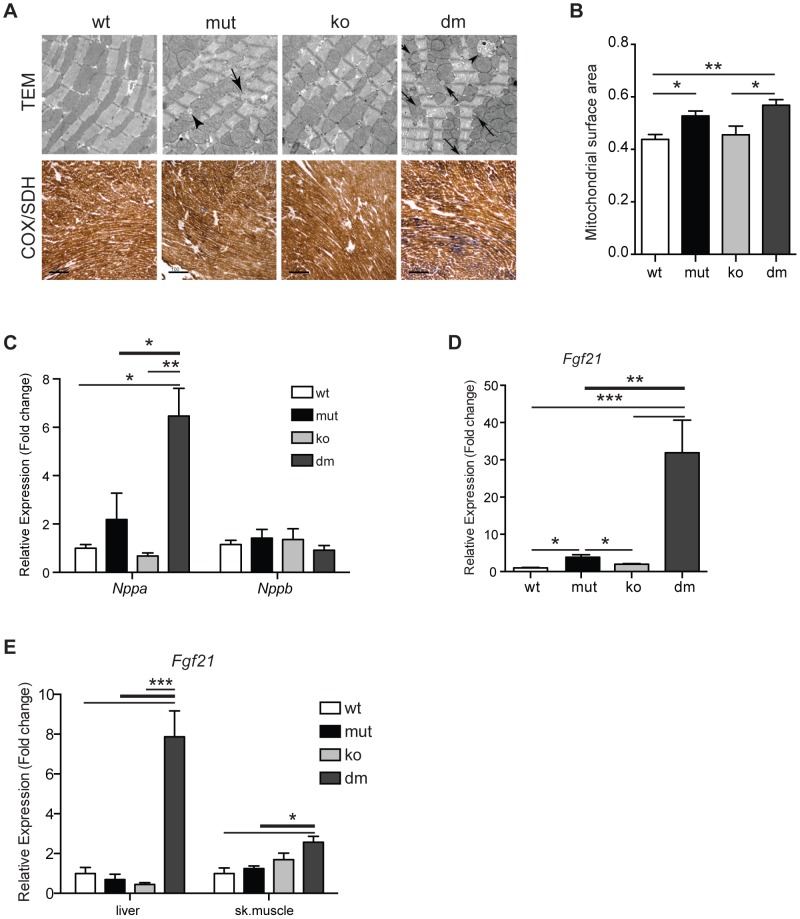
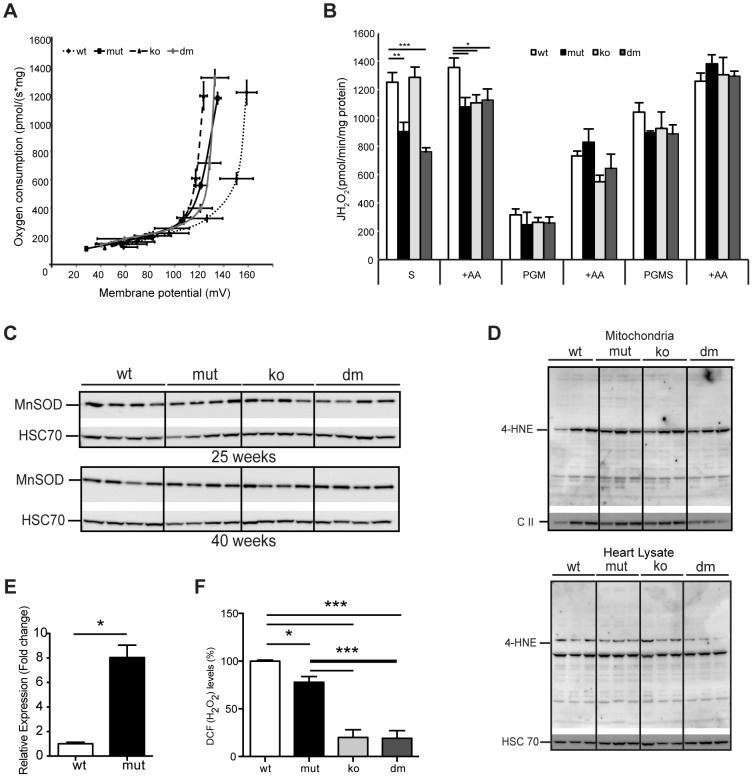
Although mitochondrial dysfunction is often accompanied by excessive reactive oxygen species (ROS) production, we previously showed that an increase in random somatic mtDNA mutations does not result in increased oxidative stress. Normal levels of ROS and oxidative stress could also be a result of an active compensatory mechanism such as a mild increase in proton leak. Uncoupling protein 2 (UCP2) was proposed to play such a role in many physiological situations. However, we show that upregulation of UCP2 in mtDNA mutator mice is not associated with altered proton leak kinetics or ROS production, challenging the current view on the role of UCP2 in energy metabolism. Instead, our results argue that high UCP2 levels allow better utilization of fatty acid oxidation resulting in a beneficial effect on mitochondrial function in heart, postponing systemic lactic acidosis and resulting in longer lifespan in these mice. This study proposes a novel mechanism for an adaptive response to mitochondrial cardiomyopathy that links changes in metabolism to amelioration of respiratory chain deficiency and longer lifespan.
尽管线粒体功能障碍常伴随着活性氧(ROS)的过量产生,但我们之前的研究表明,随机体细胞线粒体DNA(mtDNA)突变的增加并不会导致氧化应激的增加。正常水平的ROS和氧化应激也可能是一种活跃的补偿机制的结果,比如质子泄漏的轻度增加。解偶联蛋白2(UCP2)在许多生理情况下被认为发挥着这样的作用。然而,我们发现mtDNA突变小鼠中UCP2的上调与质子泄漏动力学或ROS产生的改变无关,这对目前关于UCP2在能量代谢中作用的观点提出了挑战。相反,我们的结果表明,高水平的UCP2能更好地利用脂肪酸氧化,从而对心脏的线粒体功能产生有益影响,延缓全身性乳酸酸中毒,并延长这些小鼠的寿命。本研究提出了一种针对线粒体心肌病的适应性反应的新机制,该机制将代谢变化与呼吸链缺陷的改善以及更长的寿命联系起来。