Center for Biological Sequence analysis, Technical University of Denmark, Lyngby DK-2800, Denmark.
National Food Institute, Technical University of Denmark, Søborg DK-2860, Denmark.
Microbiome. 2014 Jun 5;2:19. doi: 10.1186/2049-2618-2-19. eCollection 2014.
In recent years, studies on the human intestinal microbiota have attracted tremendous attention. Application of next generation sequencing for mapping of bacterial phylogeny and function has opened new doors to this field of research. However, little attention has been given to the effects of choice of methodology on the output resulting from such studies.
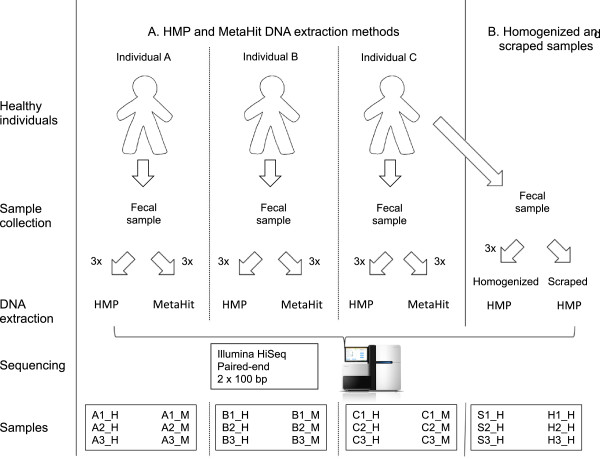
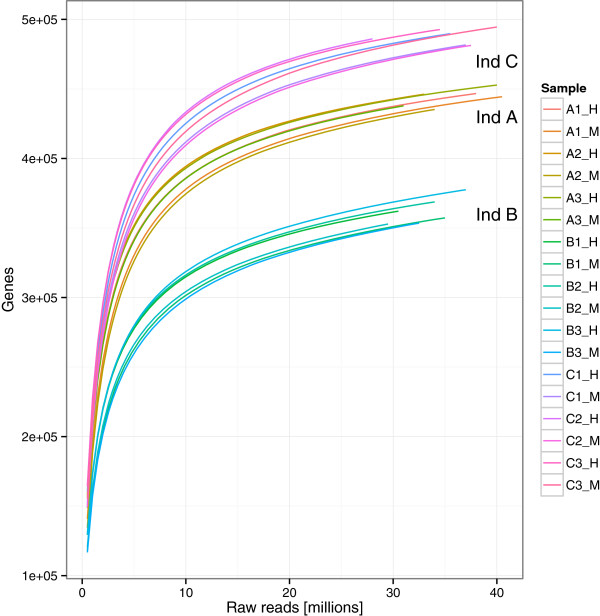
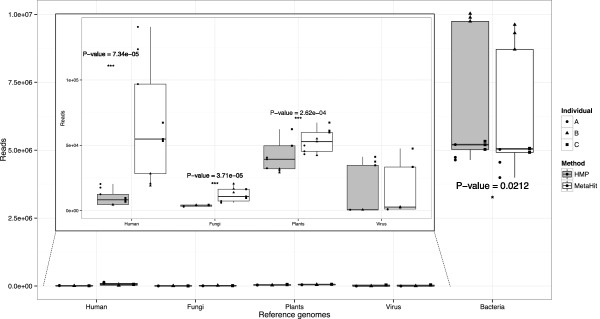
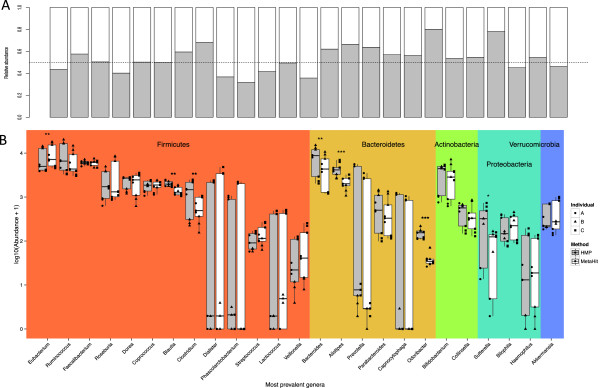
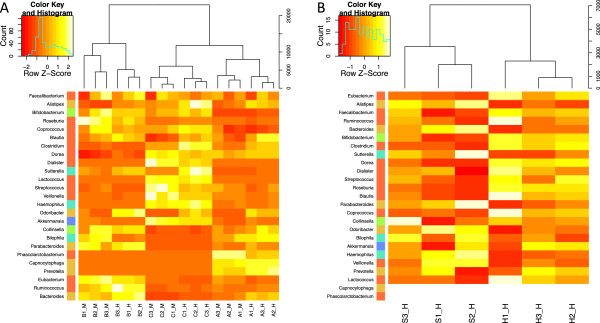
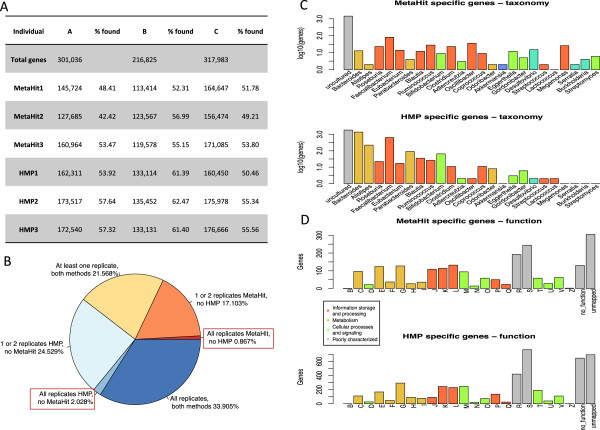
IN THIS STUDY WE CONDUCTED A SYSTEMATIC COMPARISON OF THE DNA EXTRACTION METHODS USED BY THE TWO MAJOR COLLABORATIVE EFFORTS: The European MetaHIT and the American Human Microbiome Project (HMP). Additionally, effects of homogenizing the samples before extraction were addressed. We observed significant differences in distribution of bacterial taxa depending on the method. While eukaryotic DNA was most efficiently extracted by the MetaHIT protocol, DNA from bacteria within the Bacteroidetes phylum was most efficiently extracted by the HMP protocol.
Whereas it is comforting that the inter-individual variation clearly exceeded the variation resulting from choice of extraction method, our data highlight the challenge of comparing data across studies applying different methodologies.
近年来,人类肠道微生物组的研究受到了极大的关注。应用下一代测序技术来绘制细菌系统发育和功能图谱为该研究领域开辟了新的途径。然而,对于选择的方法对研究结果的影响,人们关注甚少。
在本研究中,我们对两个主要合作项目(欧洲 MetaHIT 和美国人类微生物组计划(HMP))所使用的 DNA 提取方法进行了系统比较。此外,还研究了在提取前对样品进行均化处理的效果。我们观察到,不同的方法对细菌分类群的分布有显著的影响。虽然真核 DNA 最有效地被 MetaHIT 方案提取,但是厚壁菌门的细菌 DNA 最有效地被 HMP 方案提取。
尽管个体间的差异明显大于提取方法选择所导致的差异,令人欣慰,但我们的数据强调了在应用不同方法学的研究之间进行数据比较的挑战。