Fischer Marcus, Coleman Ryan G, Fraser James S, Shoichet Brian K
1] Department of Pharmaceutical Chemistry, University of California San Francisco, San Francisco, California 94158, USA [2] Faculty of Pharmacy, Donnelly Center, University of Toronto, 160 College St, Toronto, Ontario M5S 3E1 Canada [3].
1] Department of Pharmaceutical Chemistry, University of California San Francisco, San Francisco, California 94158, USA [2].
Nat Chem. 2014 Jul;6(7):575-83. doi: 10.1038/nchem.1954. Epub 2014 May 25.
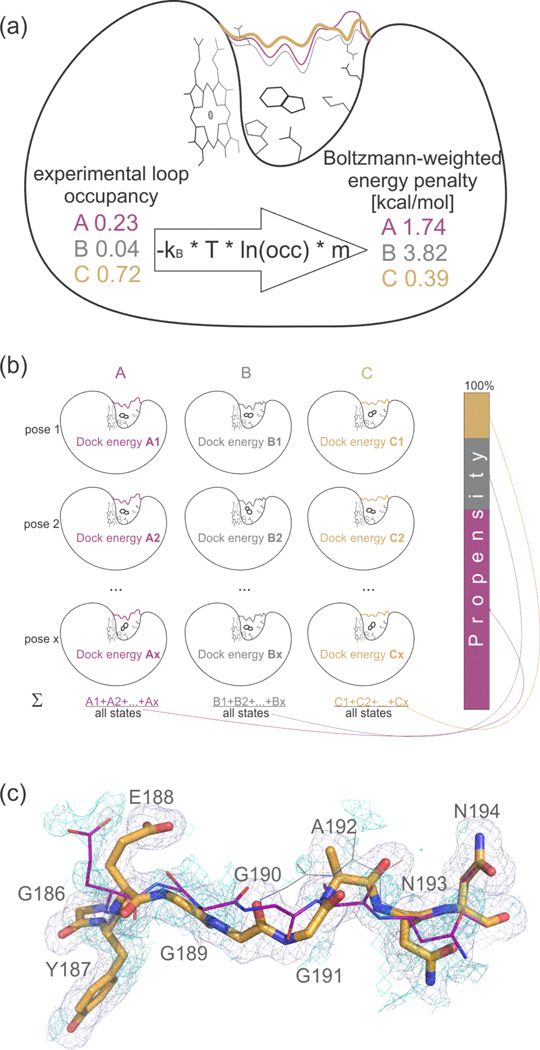
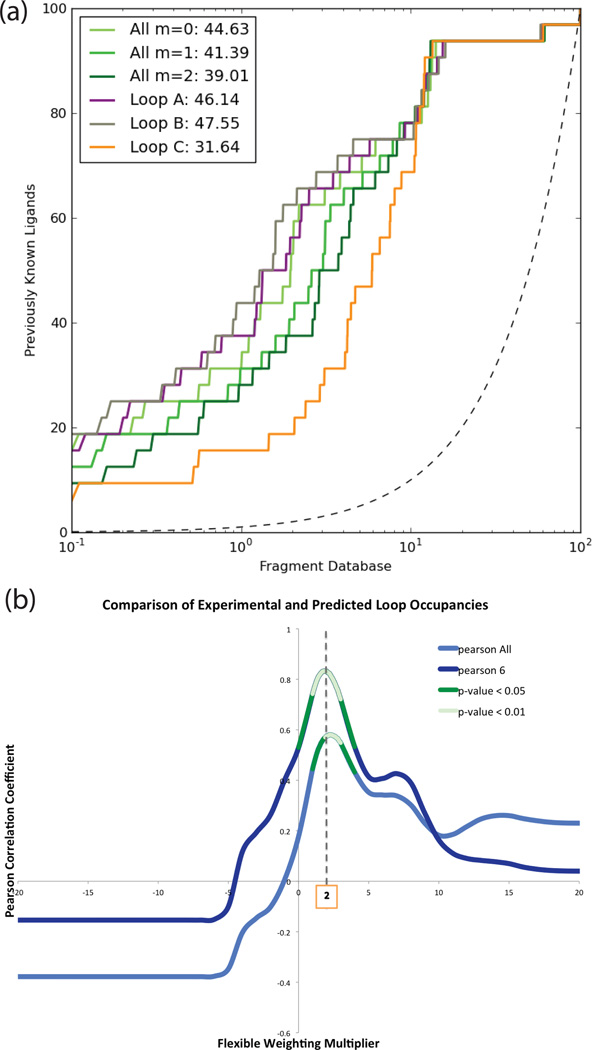
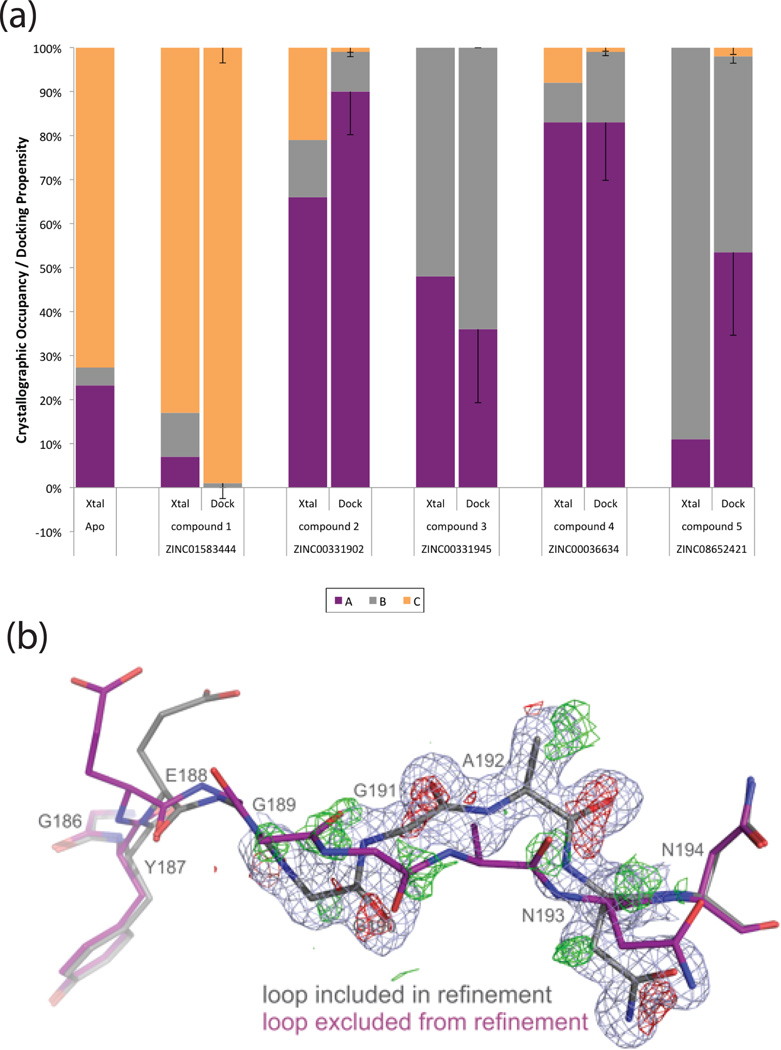
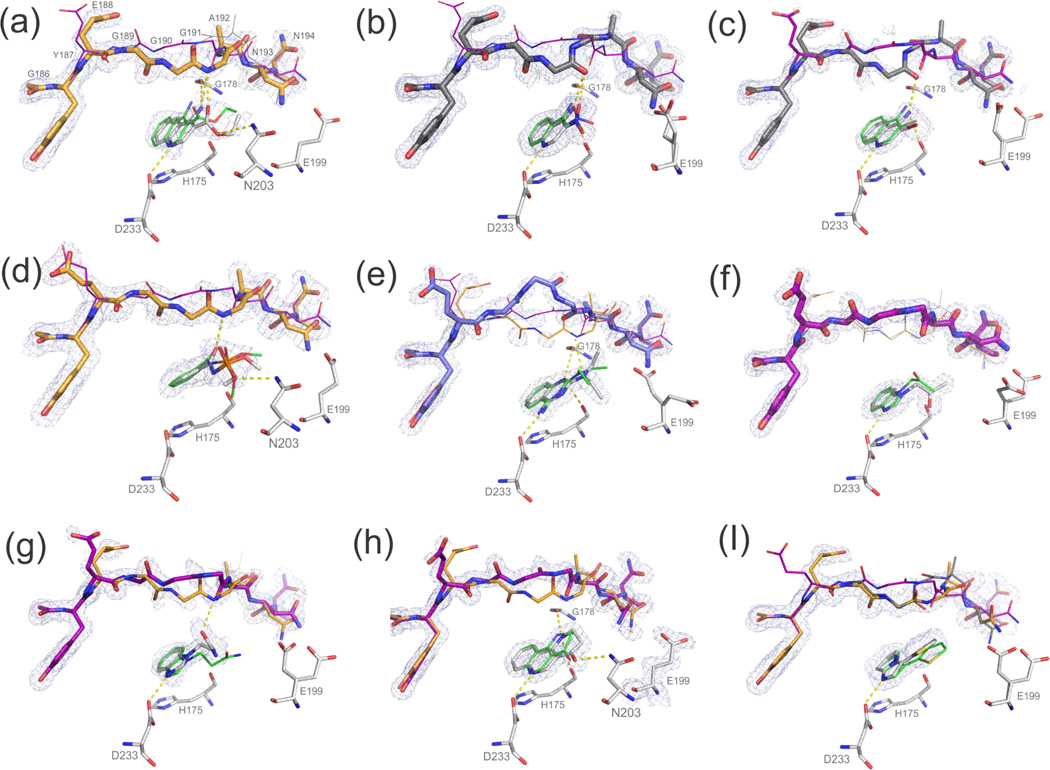
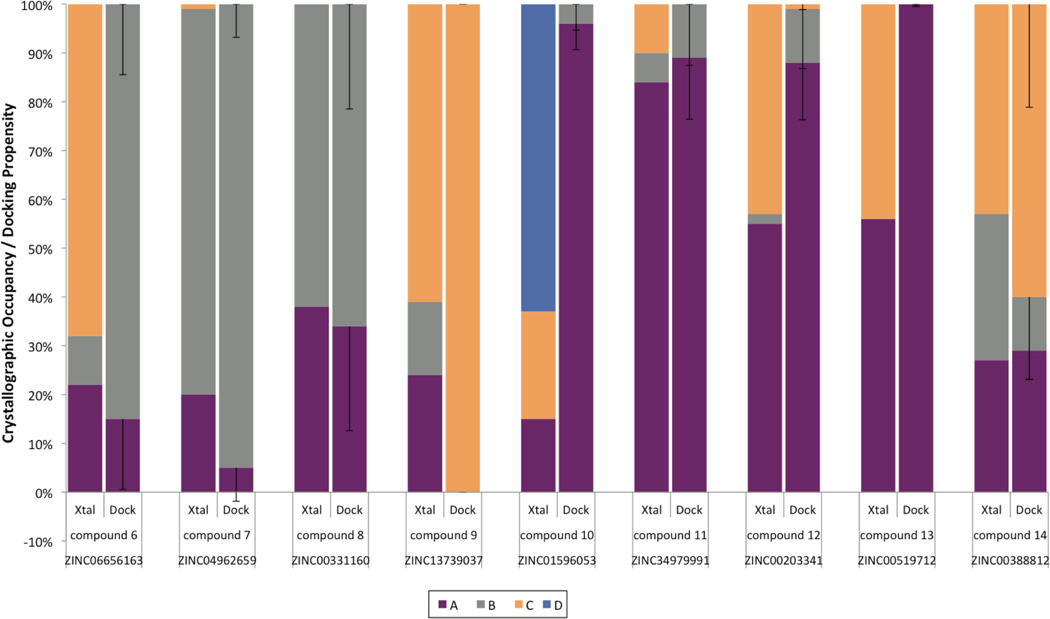
Proteins fluctuate between alternative conformations, which presents a challenge for ligand discovery because such flexibility is difficult to treat computationally owing to problems with conformational sampling and energy weighting. Here we describe a flexible docking method that samples and weights protein conformations using experimentally derived conformations as a guide. The crystallographically refined occupancies of these conformations, which are observable in an apo receptor structure, define energy penalties for docking. In a large prospective library screen, we identified new ligands that target specific receptor conformations of a cavity in cytochrome c peroxidase, and we confirm both ligand pose and associated receptor conformation predictions by crystallography. The inclusion of receptor flexibility led to ligands with new chemotypes and physical properties. By exploiting experimental measures of loop and side-chain flexibility, this method can be extended to the discovery of new ligands for hundreds of targets in the Protein Data Bank for which similar experimental information is available.
蛋白质在不同构象之间波动,这给配体发现带来了挑战,因为这种灵活性由于构象采样和能量加权问题而难以通过计算处理。在此,我们描述了一种灵活对接方法,该方法以实验得出的构象为指导对蛋白质构象进行采样和加权。这些构象在无配体受体结构中可观察到,其晶体学精修占有率定义了对接的能量惩罚。在一个大型前瞻性文库筛选中,我们鉴定出了靶向细胞色素c过氧化物酶中一个腔的特定受体构象的新配体,并通过晶体学证实了配体姿态和相关受体构象预测。受体灵活性的纳入产生了具有新化学类型和物理性质的配体。通过利用环和侧链灵活性的实验测量方法,该方法可扩展到为蛋白质数据库中数百个有类似实验信息可用的靶点发现新配体。