Ma Bing, Charkowski Amy O, Glasner Jeremy D, Perna Nicole T
Genome Center of Wisconsin, University of Wisconsin-Madison, Madison, WI 53706, USA.
BMC Genomics. 2014 Jun 21;15:508. doi: 10.1186/1471-2164-15-508.
A wealth of genome sequences has provided thousands of genes of unknown function, but identification of functions for the large numbers of hypothetical genes in phytopathogens remains a challenge that impacts all research on plant-microbe interactions. Decades of research on the molecular basis of pathogenesis focused on a limited number of factors associated with long-known host-microbe interaction systems, providing limited direction into this challenge. Computational approaches to identify virulence genes often rely on two strategies: searching for sequence similarity to known host-microbe interaction factors from other organisms, and identifying islands of genes that discriminate between pathogens of one type and closely related non-pathogens or pathogens of a different type. The former is limited to known genes, excluding vast collections of genes of unknown function found in every genome. The latter lacks specificity, since many genes in genomic islands have little to do with host-interaction.
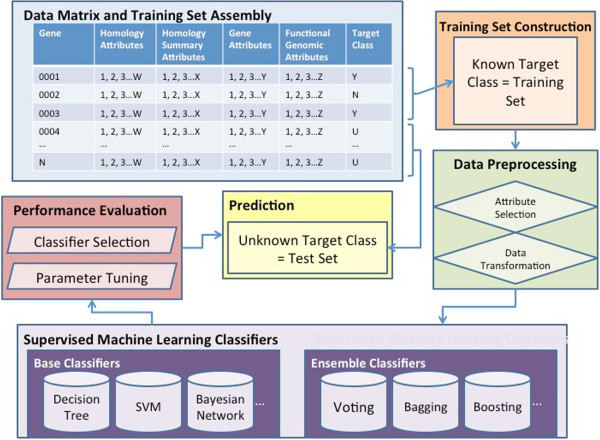
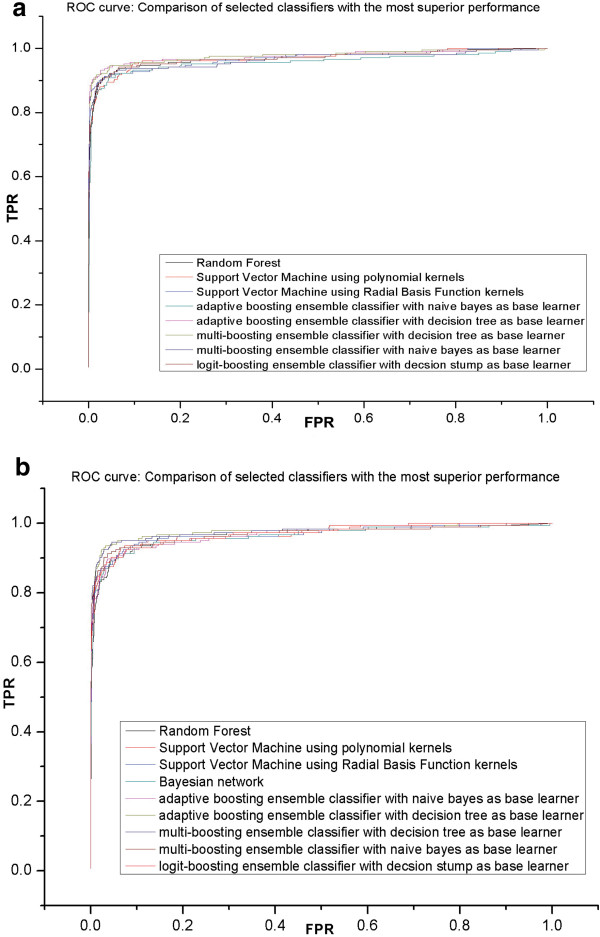
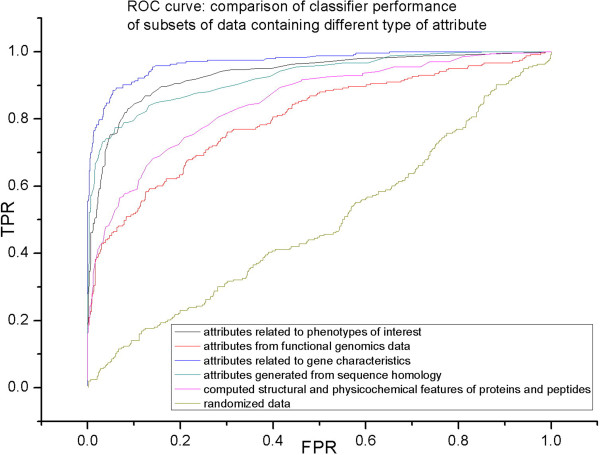
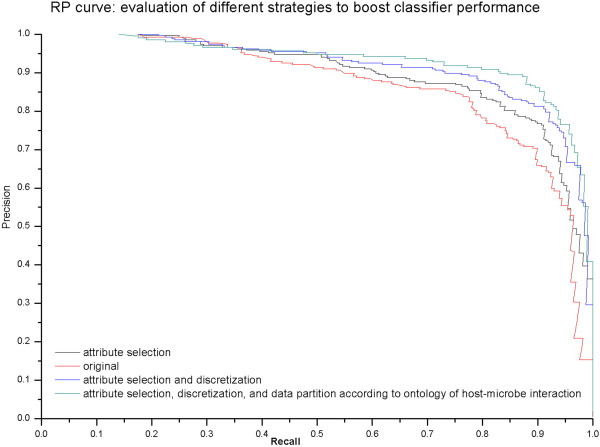
In this study, we developed a supervised machine learning approach that was designed to recognize patterns from large and disparate data types, in order to identify candidate host-microbe interaction factors. The soft rot Enterobacteriaceae strains Dickeya dadantii 3937 and Pectobacterium carotovorum WPP14 were used for development of this tool, because these pathogens are important on multiple high value crops in agriculture worldwide and more genomic and functional data is available for the Enterobacteriaceae than any other microbial family. Our approach achieved greater than 90% precision and a recall rate over 80% in 10-fold cross validation tests.
Application of the learning scheme to the complete genome of these two organisms generated a list of roughly 200 candidates, many of which were previously not implicated in plant-microbe interaction and many of which are of completely unknown function. These lists provide new targets for experimental validation and further characterization, and our approach presents a promising pattern-learning scheme that can be generalized to create a resource to study host-microbe interactions in other bacterial phytopathogens.
大量的基因组序列已提供了数千个功能未知的基因,但确定植物病原体中大量假设基因的功能仍然是一项挑战,这影响着所有关于植物-微生物相互作用的研究。数十年来对发病机制分子基础的研究集中在与长期已知的宿主-微生物相互作用系统相关的有限数量的因素上,为应对这一挑战提供的指导有限。识别毒力基因的计算方法通常依赖于两种策略:搜索与其他生物体中已知的宿主-微生物相互作用因子的序列相似性,以及识别区分一种类型的病原体与密切相关的非病原体或不同类型病原体的基因岛。前者仅限于已知基因,排除了每个基因组中发现的大量功能未知的基因集合。后者缺乏特异性,因为基因岛中的许多基因与宿主相互作用几乎无关。
在本研究中,我们开发了一种监督机器学习方法,旨在从大量不同的数据类型中识别模式,以确定候选的宿主-微生物相互作用因子。软腐肠杆菌菌株胡萝卜软腐果胶杆菌3937和胡萝卜果胶杆菌WPP14被用于开发此工具,因为这些病原体在全球农业中的多种高价值作物上具有重要影响,并且与任何其他微生物家族相比,肠杆菌科有更多的基因组和功能数据。我们的方法在10折交叉验证测试中实现了超过90%的精度和超过8%的召回率。
将该学习方案应用于这两种生物体的完整基因组产生了一份约200个候选基因的列表,其中许多以前未涉及植物-微生物相互作用,并且许多功能完全未知。这些列表为实验验证和进一步表征提供了新的靶点,并且我们的方法提出了一种有前景的模式学习方案,该方案可以推广以创建一种资源来研究其他细菌性植物病原体中的宿主-微生物相互作用。